1
PRODUCT MONOGRAPH
Pr
BARACLUDE*
(entecavir)
Tablets 0.5 mg
Antiviral
Bristol-Myers Squibb Canada Date of Preparation:
Montreal, Canada June 16, 2006
* TM of Bristol-Myers Squibb Company used under license by
Bristol-Myers Squibb Canada Date of revision:
23 September 2020
Control No. 233293
2
Table of Contents
PART I: HEALTH PROFESSIONAL INFORMATION ......................................................... 3
SUMMARY PRODUCT INFORMATION................................................................................ 3
INDICATIONS AND CLINICAL USE ..................................................................................... 3
CONTRAINDICATIONS ........................................................................................................... 3
WARNINGS AND PRECAUTIONS ......................................................................................... 3
ADVERSE REACTIONS ........................................................................................................... 6
DRUG INTERACTIONS ......................................................................................................... 10
DOSAGE AND ADMINISTRATION ..................................................................................... 11
OVERDOSAGE ........................................................................................................................ 12
ACTION AND CLINICAL PHARMACOLOGY .................................................................... 12
STORAGE AND STABILITY ................................................................................................. 19
DOSAGE FORMS, COMPOSITION AND PACKAGING..................................................... 19
PART II: SCIENTIFIC INFORMATION ............................................................................... 21
PHARMACEUTICAL INFORMATION ................................................................................. 21
CLINICAL TRIALS ................................................................................................................. 21
DETAILED PHARMACOLOGY ............................................................................................ 30
TOXICOLOGY ......................................................................................................................... 30
PART III: CONSUMER INFORMATION .............................................................................. 40
ABOUT THIS MEDICATION ................................................................................................ 40
WARNINGS AND PRECAUTIONS ....................................................................................... 40
INTERACTIONS WITH THIS MEDICATION ...................................................................... 41
PROPER USE OF THIS MEDICATION ................................................................................. 41
SIDE EFFECTS AND WHAT TO DO ABOUT THEM ......................................................... 42
HOW TO STORE IT ................................................................................................................. 42

3
Pr
BARACLUDE*
(entecavir)
PART I: HEALTH PROFESSIONAL INFORMATION
SUMMARY PRODUCT INFORMATION
Route of
Administration
Dosage Form /
Strength
Clinically Relevant Nonmedicinal Ingredients
Oral
Tablets 0.5 mg
lactose monohydrate
For a complete listing, see Dosage Forms,
Composition and Packaging section
INDICATIONS AND CLINICAL USE
BARACLUDE (entecavir) is indicated for the treatment of chronic hepatitis B virus infection in
adults with evidence of active viral replication and either evidence of persistent elevations in
serum aminotransferases (ALT or AST) or histologically active disease.
This indication is based on efficacy and safety data in nucleoside-treatment-naive and in
lamivudine-refractory adult patients with HBeAg-positive or HBeAg-negative chronic HBV
infection with compensated liver disease and on more limited data in adult patients with
HIV/HBV co-infection who have received prior lamivudine therapy.
CONTRAINDICATIONS
BARACLUDE is contraindicated in patients with previously demonstrated hypersensitivity to
entecavir or any component of the product. (For a complete listing, see Dosage Forms,
Composition and Packaging section).
WARNINGS AND PRECAUTIONS
Severe acute exacerbations of hepatitis B have been reported in patients who have
discontinued anti-hepatitis B therapy, including entecavir. Hepatic function should be
monitored closely with both clinical and laboratory follow-up for at least several months in
patients who discontinue anti-hepatitis B therapy. If appropriate, re-initiation of anti-
hepatitis B therapy may be warranted (see ADVERSE REACTIONS: Exacerbations of
Hepatitis After Discontinuation of Treatment).
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been
reported with the use of nucleoside analogues, including BARACLUDE, alone or in
combination with antiretrovirals. Patients with decompensated liver disease may be at
higher risk for lactic acidosis.
Limited clinical experience suggests there is a potential for the development of resistance to

4
HIV (human immunodeficiency virus) nucleoside reverse transcriptase inhibitors if
BARACLUDE is used to treat chronic hepatitis B virus infection in patients with HIV
infection that is not being treated. Therapy with BARACLUDE is not recommended for
HIV/HBV co-infected patients who are not also receiving highly active antiretroviral
therapy (HAART). (see WARNINGS AND PRECAUTIONS: Patients co-infected with
HIV and HBV)
General
BARACLUDE tablets contain lactose and are not recommended for patients with rare hereditary
problems of galactose intolerance, the Lapp lactase deficiency or glucose-galactose
malabsorption.
Carcinogenesis, Mutagenesis, Impairment of Fertility
Positive carcinogenic results were found in two-year carcinogenicity studies with entecavir
conducted in mice and rats. In male mice, increases in the incidences of lung adenomas were
observed at exposures ≥ 3 times the exposure in humans at 1 mg and lung carcinomas were
observed in male and female mice at approximately 40 times the exposure in humans at 1 mg.
Tumor development was preceded by pneumocyte proliferation in the lung, which was not
observed in rats, dogs, or monkeys administered entecavir, indicating that a key event in lung
tumor development observed in mice likely was species specific. Drug-related increased
incidences of other types of tumors were seen at the highest entecavir exposures [in mice
approximately 40 times and in rats 35 times (males) and 24 times (females) human exposure at 1
mg], including liver carcinomas in male mice, benign vascular tumors in female mice, brain
microglial tumors in male and female rats, and liver adenomas and carcinomas in female rats.
Skin fibromas were observed in female rats at both the high (0.4 mg/kg/day; equivalent to 4 times
the exposure in humans at 1 mg) and highest (2.6 mg/kg/day; equivalent to 24 times the exposure
in humans at 1 mg) doses. (see TOXICOLOGY, Carcinogenesis, Mutagenesis, Impairment
of Fertility for more detailed information).
It is not known how predictive the results of rodent carcinogenicity studies may be for humans.
(see CLINICAL TRIALS, Data from Long-term Observational Study).
Entecavir was clastogenic to human lymphocyte cultures and in mouse lymphoma cells in vitro.
Entecavir was not mutagenic in the Ames bacterial reverse mutation assay, a mammalian-cell
gene mutation assay, and a transformation assay with Syrian hamster embryo cells. Entecavir was
also negative in an oral micronucleus study and an oral DNA repair study in rats. In reproductive
toxicology studies in which rats were administered entecavir at up to 30 mg/kg for up to 4 weeks,
no evidence of impaired fertility was seen in males or females at systemic exposures >90 times
those in humans at 1 mg. In rodent and dog toxicology studies, seminiferous tubular degeneration
was observed at ≥ 35 times the exposure in humans at 1 mg. No testicular changes were evident
in monkeys administered entecavir for 1 year at 167 times the exposure in humans at 1 mg.
Liver Transplant Recipients
The safety and efficacy of BARACLUDE in liver transplant recipients are unknown. The
5
potential for pharmacokinetic interaction between entecavir and the immunosuppressants
cyclosporine A or tacrolimus was not formally evaluated. If BARACLUDE treatment is
determined to be necessary for a liver transplant recipient who has received or is receiving
cyclosporine or tacrolimus, renal function must be carefully monitored both before and during
treatment with BARACLUDE (see ACTION AND CLINICAL PHARMACOLOGY: Special
Populations and DOSAGE AND ADMINISTRATION: Renal Impairment).
Renal Impairment
BARACLUDE is predominantly eliminated by the kidney. Dosage adjustment of BARACLUDE
is recommended for patients with a creatinine clearance <50 mL/min, including patients on
hemodialysis or CAPD [continuous ambulatory peritoneal dialysis] (see DOSAGE AND
ADMINISTRATION: Renal Impairment).
Special Populations
Patients co-infected with HIV and HBV
BARACLUDE has not been evaluated in patients who are co-infected with HIV and HBV and
are not concurrently receiving effective HIV treatment. Limited clinical experience suggests
there is a potential for the development of resistance to HIV nucleoside reverse transcriptase
inhibitors if BARACLUDE is used to treat chronic hepatitis B virus infection in patients with
HIV infection that is not being treated. Therefore therapy with BARACLUDE is not
recommended for HIV/HBV co-infected patients who are not also receiving highly active
antiretroviral therapy (HAART). BARACLUDE has not been studied as a treatment for HIV
infection and is not recommended for this use. (See ACTION AND CLINICAL
PHARMACOLOGY: Special Populations and Conditions, Patients Co-infected with HIV and
HBV and CLINICAL TRIALS: Special Populations, Patients Co-infected with HIV and HBV).
Before initiating BARACLUDE therapy, HIV antibody testing should be offered to all patients.
Pregnant Women
There are no adequate and well-controlled studies in pregnant women. BARACLUDE should be
used during pregnancy only if the potential benefit justifies the potential risk to the fetus.
Entecavir caused effects on embryo-fetal development in rats at doses that also produced
maternal toxicity; at these doses, exposures to entecavir were 180 times those in humans at 1 mg.
In rabbits, embryo-fetal toxicity was observed at exposures to entecavir 883 times those in
humans at 1 mg. There were no adverse effects on growth, development, and reproductive
performance in the progeny of rats administered entecavir at doses associated with exposures to
entecavir > 94 times those in humans at 1 mg. (see TOXICOLOGY, Reproductive Toxicology
for more detailed information).
Pregnancy Registry:
To monitor maternal-fetal outcomes of pregnant women exposed to BARACLUDE a Pregnancy
Registry has been established. To register patients, physicians must obtain prior consent.
6
Physicians can register patients by calling 1-800-258-4263.
Labor and Delivery
There are no studies in pregnant women and no data on the effect of BARACLUDE on
transmission of HBV from mother to infant. Therefore, appropriate interventions should be used
to prevent neonatal acquisition of HBV.
Nursing Women
Entecavir is excreted in the milk of rats. It is not known whether this drug is excreted in human
milk. Mothers should be instructed not to breast-feed if they are taking BARACLUDE.
Pediatrics (<16 years of age)
Safety and effectiveness of BARACLUDE in pediatric patients below the age of 16 years have
not been established.
Geriatrics (>65 years of age)
Clinical studies of BARACLUDE did not include sufficient numbers of subjects aged 65 years
and over to determine whether they respond differently from younger subjects. Entecavir is
substantially excreted by the kidney, and the risk of toxic reactions to this drug may be greater in
patients with impaired renal function. Because elderly patients are more likely to have decreased
renal function, care should be taken in dose selection, and it may be useful to monitor renal
function (see DOSAGE AND ADMINISTRATION: Renal Impairment).
Use in Racial/Ethnic Groups
Clinical studies of BARACLUDE did not include sufficient numbers of subjects from some
racial/ethnic minorities (black/African American, Hispanic) to determine whether they respond
differently to treatment with the drug. There are no significant racial differences in entecavir
pharmacokinetics.
ADVERSE REACTIONS
Adverse Drug Reaction Overview
Assessment of adverse reactions is based on four pivotal studies (AI463014, AI463022,
AI463026, and AI463027) in which 1720 patients with chronic hepatitis B infection received
double-blind treatment with BARACLUDE 0.5 mg/day (n=679), BARACLUDE 1 mg/day
(n=183), or lamivudine (n=858) for up to two years (Studies AI463022, AI463027 for
nucleoside-naïve patients and studies AI463014, AI463026 for lamivudine-refractory patients).
The safety profiles of BARACLUDE and lamivudine were comparable in these studies.
The safety profile of BARACLUDE 1 mg (n=51) in HIV/HBV co-infected patients enrolled in
Study AI463038 was similar to that of placebo (n=17) through 24 weeks of blinded treatment and
7
similar to that seen in non-HIV infected patients. (See WARNINGS AND PRECAUTIONS –
Special Populations: Patients Co-infected with HIV and HBV)
The most common (≥ 3%) adverse events of any severity with at least a possible relation to study
drug for BARACLUDE-treated patients were headache, fatigue, dizziness, and nausea. The most
common adverse events among lamivudine-treated patients were headache, fatigue, and
dizziness. One percent of BARACLUDE-treated patients in these four studies compared with 4%
of lamivudine-treated patients discontinued for adverse events or abnormal laboratory test results.
Clinical Trial Adverse Drug Reactions
Because clinical trials are conducted under very specific conditions the adverse reaction rates
observed in the clinical trials may not reflect the rates observed in practice and should not be
compared to the rates in the clinical trials of another drug. Adverse drug reaction information
from clinical trials is useful for identifying drug-related adverse events and for approximating
rates.
Clinical adverse reactions occurring in ≥ 3% of BARACLUDE-treated patients during therapy in
four clinical studies in which BARACLUDE was compared with lamivudine, in addition to
selected clinical adverse reactions that occurred in < 3% of patients are presented in Table 1.
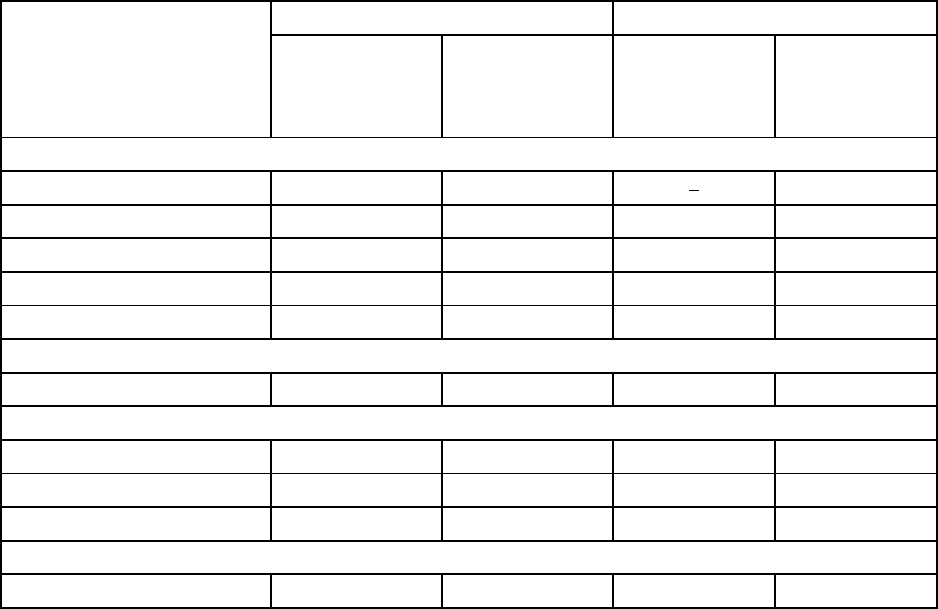
8
Table 1:Clinical Adverse Reactions Reported in ≥ 3% of BARACLUDE-treated Patients,
Plus Selected Clinical Adverse Reactions in Four BARACLUDE Clinical Trials – Through
2 Years of Treatment
Body System/
Adverse Event
a
Nucleoside-Naive
b
Lamivudine-Refractory
c
BARACLUDE
0.5 mg
n = 679
%
Lamivudine
100 mg
n = 668
%
BARACLUDE
1 mg
n = 183
%
Lamivudine
100 mg
n = 190
%
Gastrointestinal
Nausea
3
2
4
3
Abdominal pain upper
3
2
2
5
Dyspepsia
2
2
3
<1
Diarrhea
1
<1
2
1
Vomiting
1
<1
1
<1
General
Fatigue
5
5
9
6
Nervous System
Headache
8
8
10
7
Dizziness
4
3
5
2
Somnolence
1
1
2
1
Psychiatric
Insomnia
2
1
1
<1
a Includes events of possible, probable, certain, or unknown relationship to treatment regimen.
b
Studies AI463022 and AI463027 Mean duration of therapy was 69 weeks for BARACLUDE-treated and 63
weeks for lamivudine-treated patients.
c
Includes Study AI463026 and the BARACLUDE 1-mg and lamivudine treatment arms of Study
AI463014, a Phase 2 multinational, randomized, double-blind study of three doses of BARACLUDE (0.1, 0.5, and 1
mg) once daily versus continued lamivudine 100 mg once daily for up to 52 weeks in patients who experienced
recurrent viremia on lamivudine therapy. Mean duration of therapy was 73 weeks for BARACLUDE-treated and 51
weeks for lamivudine-treated patients.
Exacerbations of Hepatitis After Discontinuation of Treatment
In the Phase 3 studies, a subset of patients was allowed to discontinue treatment at or after 52
weeks if they achieved a protocol-defined response to therapy. An exacerbation of hepatitis or
ALT flare was defined as ALT >10 X ULN and >2 X the patient’s reference level (minimum of
the baseline or last measurement at end of dosing). As demonstrated in Table 2, a proportion of
patients experienced post-treatment ALT flares. If BARACLUDE is discontinued without regard
to treatment response, the rate of post-treatment flares could be higher.
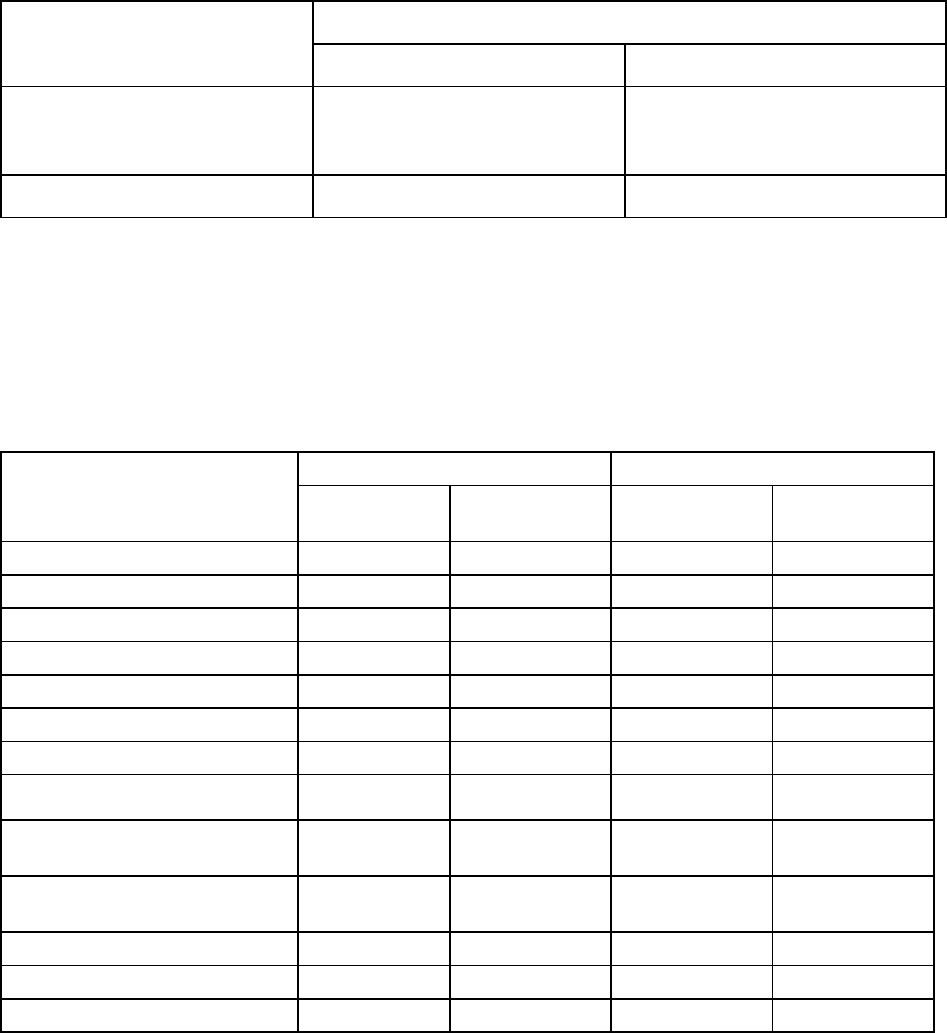
9
Table 2: Exacerbations of Hepatitis During Off-Treatment Follow-up, Patients in Studies
AI463022 , AI463027 and AI463026
Patients with ALT Elevations > 10xULN and >2 x Reference
a
BARACLUDE
Lamivudine
Total Nucleoside-naïve
HBeAg-positive
HBeAg-negative
28/476 (6%)
4/174 (2%)
24/302 (8%)
43/417 (10%)
13/147 (9%)
30/270 (11%)
Lamivudine-refractory
6/52 (12%)
0/16
a
Reference is the minimum of the baseline or last measurement at end of dosing. Median time to off-treatment
exacerbation was 23 weeks for BARACLUDE-treated patients and 10 weeks for lamivudine-treated patients.
Abnormal Hematologic and Clinical Chemistry Findings
Frequencies of selected treatment-emergent laboratory abnormalities reported during therapy in
four clinical trials of BARACLUDE compared with lamivudine are listed in Table 3.
Table 3: Selected Treatment-Emergent
a
Laboratory Abnormalities Reported in Four
BARACLUDE Clinical Trials -Through 2 Years
Test
Nucleoside -Naive
b
Lamivudine-Refractory
c
BARACLUDE
0.5 mg (n = 679)
Lamivudine
100 mg (n = 668)
BARACLUDE
1 mg (n = 183)
Lamivudine
100 mg (n = 190)
ALT> 10 x ULN and > 2 x baseline
2%
4%
2%
11%
ALT > 5.0 ULN
11%
16%
12%
24%
AST > 5.0 ULN
5%
8%
5%
17%
Albumin < 2.5 g/dL
<1%
<1%
0%
2%
Total bilirubin > 2.5 x ULN
2%
2%
3%
2%
Amylase > 2.1 x ULN
2%
2%
3%
3%
Lipase > 2.1 x ULN
7%
6%
7%
7%
Creatinine ≻ 3.0 x ULN
0% 0% 0% 0%
Confirmed creatinine increase ≥
44.2 mmol/L
1% 1% 2% 1%
Hyperglycemia fasting
>13.8 mmol/L
2% 1% 3% 1%
Glycosuria
d
4%
3%
4%
6%
Hematuria
e
9%
10%
9%
6%
Platelets < 50,000/mm
3
<1%
<1%
<1%
<1%
a
On-treatment value worsened from baseline to Grade 3 or Grade 4 for all parameters except albumin (any on treatment
value <2.5 g/dL), confirmed creatinine increase ≥ 44.2 mmol/L and ALT >10 X ULN and >2 X baseline.
b
Studies AI463022 and AI463027. . Mean duration of therapy was 69 weeks for BARACLUDE-treated and 63 weeks
for lamivudine-treated patients.
c
Includes Study AI463026 and the BARACLUDE 1-mg and lamivudine treatment arms of Study AI463014, a Phase 2
multinational, randomized, double-blind study of three doses of BARACLUDE (0.1, 0.5, and 1 mg) once daily versus
continued lamivudine 100 mg once daily for up to 52 weeks in patients who experienced recurrent viremia on
lamivudine therapy. Mean duration of therapy was 73 weeks for BARACLUDE-treated and 51 weeks for lamivudine-
treated patients.
d
Grade 3=3+, large, ≥ 500 mg/dL; Grade 4=4+, marked, severe
e
Grade 3=3+, large, Grade 4 =
≥ 4+, marked, severe, many
ULN= upper limit of normal
10
Among BARACLUDE-treated patients in these studies, on-treatment ALT elevations >10 X
ULN and >2 X baseline generally resolved with continued treatment. A majority of these
exacerbations were associated with a ≥ 2 log
10
/mL reduction in viral load that preceded or
coincided with the ALT elevation. Periodic monitoring of hepatic function is recommended
during treatment.
Post-Market Adverse Drug Reactions
The following events have been identified during postapproval use of BARACLUDE. Because
reports are voluntary from a population of unknown size, an estimate of frequency cannot be
made, as well, the existence of underlying medical conditions confounds the assessment of
causality.
Gastrointestinal disorders: upper abdominal pain, pancreatitis
Metabolism and nutrition disorders: lactose intolerance. Lactic acidosis has been reported, often
in association with hepatic decompensation, other serious medical conditions, or drug exposures.
Patients with decompensated liver disease may be at higher risk for lactic acidosis.
Hepatobiliary disorders: increased transaminases
Skin and subcutaneous tissue disorders: alopecia, rash
Blood and lymphatic system disorders: leukopenia, neutropenia, platelet count decreased
Immune System Disorders: hypersensitivity and drug hypersensitivity including anaphylactoid
reaction
DRUG INTERACTIONS
Overview
Since entecavir is primarily eliminated by the kidneys (see ACTION AND CLINICAL
PHARMACOLOGY: Metabolism and Elimination), coadministration of BARACLUDE with
drugs that reduce renal function or compete for active tubular secretion may increase serum
concentrations of either entecavir or the coadministered drug. In clinical trials, coadministration
of BARACLUDE with lamivudine, adefovir dipivoxil, or tenofovir disoproxil fumarate did not
result in significant drug interactions. The effects of coadministration of BARACLUDE with
other drugs that are renally eliminated or are known to affect renal function have not been
evaluated, and patients should be monitored closely for adverse events when BARACLUDE is
coadministered with such drugs.
The metabolism of entecavir was evaluated in in vitro and in vivo studies. Entecavir is not a
substrate, inhibitor, or inducer of the cytochrome P450 (CYP450) enzyme system. At
concentrations up to approximately 10,000-fold higher than those obtained in humans, entecavir
inhibited none of the major human CYP450 enzymes 1A2, 2C9, 2C19, 2D6, 3A4, 2B6, and 2E1.
At concentrations up to approximately 340-fold higher than those observed in humans, entecavir
did not induce the human CYP450 enzymes 1A2, 2C9, 2C19, 3A4, 3A5, and 2B6. (See
ACTION AND CLINICAL PHARMACOLOGY: Metabolism and Elimination.) The
11
pharmacokinetics of entecavir are unlikely to be affected by coadministration with agents that are
either metabolized by, inhibit, or induce the CYP450 system. Likewise, the pharmacokinetics of
known CYP substrates are unlikely to be affected by coadministration of BARACLUDE.
Drug-Drug Interactions
In clinical studies, the steady-state pharmacokinetics of BARACLUDE and coadministered drug
were not altered in interaction studies of entecavir with lamivudine, adefovir dipivoxil, and
tenofovir disoproxil fumarate.
Drug-Food Interactions
Oral administration of 0.5 mg of BARACLUDE with a standard high-fat meal (945 kcal, 54.6 g
fat) or a light meal (379 kcal, 8.2 g fat) resulted in a minimal delay in absorption (1.0-1.5 hour
fed vs. 0.75 hours fasted), a decrease in C
max
of 44%-46%, and a decrease in AUC of 18%-20%.
Therefore, BARACLUDE should be administered on an empty stomach (at least 2 hours after a
meal and at least 2 hours before the next meal).
DOSAGE AND ADMINISTRATION
Recommended Dose and Dosage Adjustment
The usual recommended dose of BARACLUDE for chronic hepatitis B virus infection in adults
and adolescents 16 years of age or older is 0.5 mg once daily.
For adults and adolescents 16 years of age or older with a history of hepatitis B viremia while
receiving lamivudine or with known lamivudine resistance mutations, the recommended dose of
BARACLUDE is 1 mg (two 0.5 mg tablets) once daily.
BARACLUDE should be administered on an empty stomach (at least 2 hours after a meal and at
least 2 hours before the next meal).
Renal Impairment
In patients with renal impairment, the apparent oral clearance of entecavir decreased as creatinine
clearance decreased. Dosage adjustment is recommended for patients with creatinine clearance
<50 mL/min, including patients on hemodialysis or CAPD (continuous ambulatory peritoneal
dialysis), as shown in Table 4.
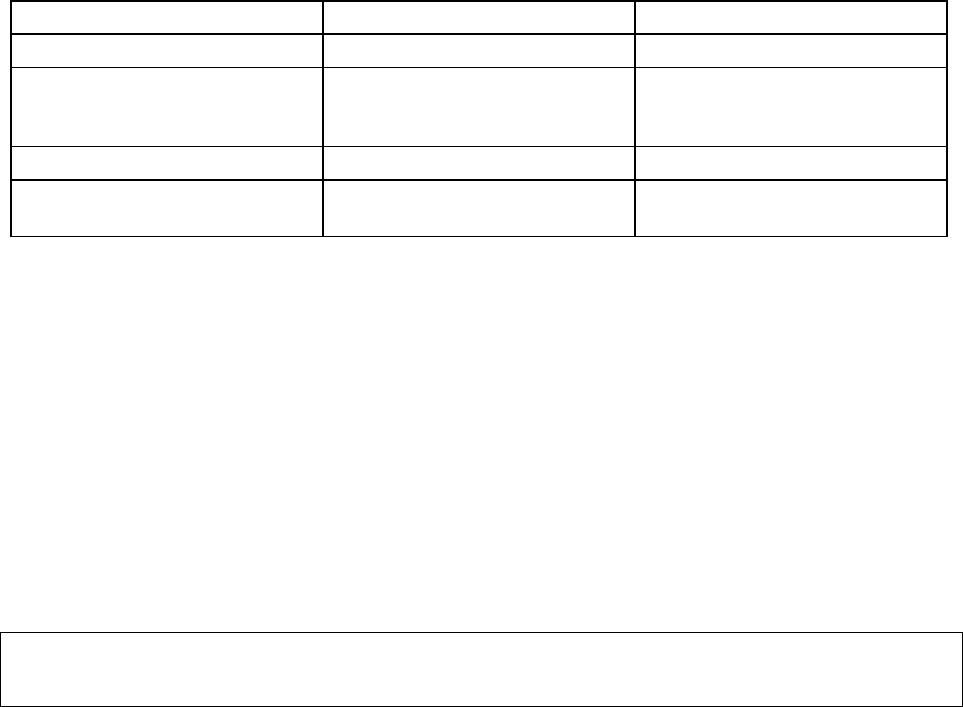
12
Table 4: Recommended Dosage of BARACLUDE in Patients with Renal Impairment
Creatinine Clearance (mL/min)
Usual Dose (0.5 mg)
Lamivudine Refractory (1 mg)
≥ 50
0.5 mg once daily
1 mg once daily
30 to <50 0.5 mg every 48 hours
0.5 mg once daily
OR
1 mg every 48 hours
10 to <30
0.5 mg every 72 hours
1 mg every 72 hours
<10
Hemodialysis
a
or CAPD
0.5 mg every 7 days 01 mg every 7 days
a
On hemodialysis days, administer after hemodialysis.
Hepatic Impairment
No dosage adjustment is necessary for patients with hepatic impairment.
Duration of Therapy
The optimal duration of treatment with BARACLUDE for patients with chronic hepatitis B
infection and the relationship between treatment and long-term outcomes such as cirrhosis and
hepatocellular carcinoma are unknown.
OVERDOSAGE
For management of a suspected drug overdose, please contact your regional Poison Control
Centre.
Activated charcoal may be administered to aid in the removal of unabsorbed drug. General
supportive measures are recommended. Healthy subjects who received single entecavir doses up
to 40 mg or multiple doses up to 20 mg/day for up to 14 days had no increase in, or unexpected,
adverse events. If overdose occurs, the patient must be monitored for evidence of toxicity, and
standard supportive treatment applied as necessary.
Following a single 1-mg dose of entecavir, a 4-hour hemodialysis session removed approximately
13% of the entecavir dose.
ACTION AND CLINICAL PHARMACOLOGY
Mechanism of Action
Entecavir is a guanosine nucleoside analogue that is efficiently phosphorylated to the active
triphosphate form and exhibits selective activity against HBV polymerase, competes with the
natural substrate deoxyguanosine triphosphate, and inhibits all three functional activities of the
HBV polymerase (reverse transcriptase, rt): (1) base priming, (2) reverse transcription of the
negative strand from the pregenomic messenger RNA, and (3) synthesis of the positive strand of
HBV DNA. Entecavir triphosphate has an inhibition constant (Ki) for HBV DNA polymerase of
0.0012 μM and is a weak inhibitor of cellular DNA polymerases α, ß, and δ and mitochondrial
13
DNA polymerase γ with Ki values ranging from 18 to >160 μM.
Antiviral Activity
Entecavir inhibited HBV DNA synthesis (50% reduction, EC50) at a concentration of 0.004 μM
in human HepG2 cells transfected with wild-type HBV. The median EC50 value for entecavir
against lamivudine resistant HBV (rtL180M, rtM204V) was 0.026 μM (range 0.010-0.059 μM).
A comprehensive analysis of the inhibitory activity of entecavir against a panel of laboratory and
clinical HIV-1 isolates using a variety of cells and assay conditions yielded EC50 values ranging
from 0.026 to >10 µM: the lower EC50 values were observed when decreased levels of virus
were used in the assay. In cell culture, entecavir selected for an M184I substitution in HIV
reverse transcriptase at micromolar concentrations, confirming inhibitory pressure at high
entecavir concentrations. HIV variants containing the M184I substitution showed loss of
susceptibility to entecavir.
The coadministration of HIV nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) with
BARACLUDE is unlikely to reduce the antiviral efficacy of BARACLUDE against HBV or of
any of these agents against HIV. In HBV combination assays in vitro, abacavir, didanosine,
lamivudine, stavudine, tenofovir, or zidovudine were not antagonistic to the anti-HBV activity of
entecavir over a wide range of concentrations. In HIV antiviral assays, entecavir was not
antagonistic to the in vitro anti-HIV activity of these six NRTIs or emtricitabine at concentrations
greater than 100 times the Cmax of entecavir using the 1-mg dose.
Drug Resistance
Clinical Studies
In nucleoside-naive studies (AI463022, AI463027, and rollover study AI463901) and in studies
of lamivudine-refractory HBV (AI463026, AI463014, AI463015, and rollover study AI463901),
patients initially treated with entecavir 0.5 mg (nucleoside-naïve) or 1.0 mg (lamivudine
refractory) and with an on-therapy PCR HBV DNA measurement at or after Week 24 were
monitored for resistance. Virologic breakthroughs due to resistance to entecavir are observed in
viruses which harbour primary lamivudine resistance substitutions (M204I/V ± L180M) along
with additional substitutions at residues T184, S202 or M250 of the viral polymerase.
Nucleoside-naïve patients:
Through Year 5, genotypic evidence of entecavir resistance (ETVr) substitutions at residues
T184, S202 or M250 was observed in 3 patients (<1%), 2 of whom experienced virologic
breakthrough (see Table 5). The results reflect use of a 1 mg dose of entecavir in 147 patients in
Year 3 and all patients in Years 4 and 5 and of entecavir-lamivudine combination therapy
(followed by long-term entecavir monotherapy) for a median of 20 weeks for 130 patients in
Year 3 and for 1 week for one patient in Year 4 in a rollover study.

14
Table 5: Genotypic Entecavir Resistance and Virologic Breakthrough with Resistance
Through Year 5, Nucleoside-Naïve Studies
Year 1
Year 2
Year 3
a
Year 4
a
Year 5
a
Subjects treated and monitored for
resistance
b
663
278
149
121
108
Emerging Genotypic ETV
c,d
1
1
1
0
0
Genotypic ETVr
c,d
withVirologic
breakthrough
e
1
0
1
0
0
Cumulative probability of emerging
genotypic ETVr
c,d
0.2%
0.5%
1.2%
1.2%
1.2%
Cumulative probability of genotypic
ETVr
c,d
with virologic
breakthrough
e
0.2%
0.2%
0.8%
0.8%
0.8%
a
Results reflect the use of a 1-mg dose of entecavir for 147 subjects in Year 3 and all subjects in Years 4 and 5 and of
combination entecavir-lamivudine therapy (followed by long-term entecavir therapy) for a median of 20 weeks for 130 subjects
in Year 3 and for 1 week for 1 subject in Year 4 in rollover study.
b
Includes subjects with at least one on-therapy HBV DNA measurement by PCR at or after week 24 through week 58 (Year 1),
after week 58 through week 102 (Year 2), after week 102 through week 156 (Year 3), after week 156 through week 204 (Year 4),
or after week 204 through week 252 (Year 5).
c
ETVr = entecavir resistance substitutions at residues T184, S202 or M250.
d
Subjects also had lamivudine resistance substitutions (rtM204V and rtL180M).
e
≥1 log
10
increase above nadir in HBV DNA by PCR, confirmed with successive measurements or at the end of the windowed
point.
Emerging amino acid substitutions at M204I/V ± L180M, L80I, or V173L, which conferred
decreased phenotypic susceptibility to entecavir in the absence of S202, T184, or M250 changes,
were detected in the HBV of 3 patients (3/663 = <1%) who experienced virologic breakthrough
by the end of Year 5.
Lamivudine-refractory patients:
Through Year 5, genotypic evidence of entecavir resistance (ETVr) substitutions at residues
T184, S202 or M250 was observed in 47 patients, 39 of whom experienced virologic
breakthrough (see Table 6) The results reflect the use of entecavir-lamivudine combination
therapy (followed by long-term entecavir monotherapy) for a median of 13 weeks for 48 patients
in Year 3, for a median of 38 weeks for 10 patients in Year 4 and for 16 weeks for 1 patient in
Year 5 in a rollover study.

15
Table 6: Genotypic Entecavir Resistance and Virologic Breakthrough with Resistance
Through Year 5, Lamivudine-Refractory Studies
Year 1
Year 2
Year 3
a
Year 4
a
Year 5
a
Subjects treated and monitored for
resistance
b
187
146
80
52
33
Emerging Genotypic ETV
c,d
11
12
16
6
2
Genotypic ETVr
c,d
with Virologic
breakthrough
e
2
f
14
f
13
f
9
f
1
f
Cumulative probability of emerging
genotypic ETVr
c,d
6%
15%
36%
47%
51%
Cumulative probability of
genotypic ETVr
c,d
with virologic
breakthrough
e
1%
f
11%
f
27%
f
41%
f
44%
f
a
Results reflect the use of combination entecavir-lamivudine therapy (followed by long-term entecavir therapy) for a median of 13
weeks for 48 subjects in Year 3, for a median of 38 weeks for 10 subjects in Year 4, and for 16 weeks for 1 patient in Year 5 in a
rollover study.
b
Includes subjects with at least one on-therapy HBV DNA measurement by PCR at or after week 24 through week 58 (Year 1),
after week 58 through week 102 (Year 2), after week 102 through week 156 (Year 3) after week 156 through week 204 (Year 4),
or after week 204 through week 252 (Year 5).
c
ETVr = entecavir resistance substitutions at residues T184, S202 or M250.
d
Subjects also had lamivudine resistance substitutions (rtM204V/I± rtL180M).
e
≥1 log
10
increase above nadir in HBV DNA by PCR, confirmed with successive measurements or at the end of the windowed
point.
f
ETVr occurring in any year, virologic breakthrough in a specified year.
The presence of ETVr substitutions at baseline in isolates from 10 (5%) of 187 lamivudine-
refractory patients indicates that prior lamivudine treatment can select these resistance
substitutions and they can exist at a low frequency before entecavir treatment. Through Year 5, 3
of the 10 patients experienced virologic breakthrough. Isolates from patients who experienced
virologic breakthrough with the emergence of S202, T184 and/or M250 substitutions (n=39), had
a median 285 fold-change in entecavir susceptibility as compared to wild type HBV. Three
additional subjects experienced virologic breakthrough with the emergence of M204I/V ±
L180M, L80V or V173L/M alone.
Integrated Analysis of Phase 2 and 3 Clinical Studies
In a post-approval integrated analysis of entecavir resistance data from 17 Phase 2 and 3 clinical
studies, an emergent entecavir resistance-associated substitution rtA181C was detected in 5 out of
1461 subjects during treatment with entecavir. This substitution was detected only in the presence
of lamivudine resistance-associated substitutions rtL180M plus rtM204V.
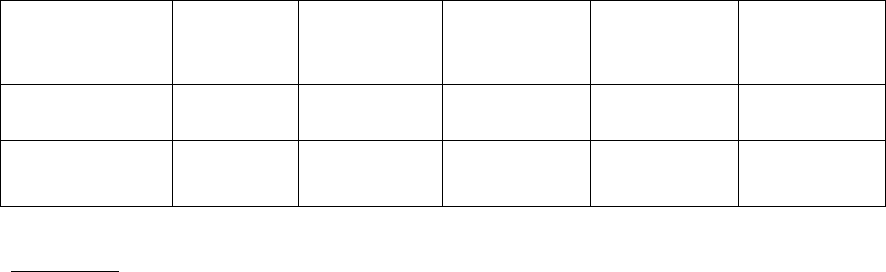
16
Cross-resistance
Cross-resistance has been observed among HBV nucleoside analogues. In cell-based assays,
HBV containing lamivudine resistance substitutions M204V/I +/- L180M was 8-fold less
susceptible to entecavir than wild type virus. Further reductions (>70 fold) in entecavir
phenotypic susceptibility required the presence of primary lamivudine resistance amino acid
substitutions (M204V/I +/- L180M ) along with additional substitutions at residues rtT184,
rtS202, or rtM250, or a combination of these substitutions with or without an rtI169 substitution
in the HBV polymerase.
Recombinant HBV genomes encoding adefovir resistance-associated substitutions at either
rtN236T or rtA181V remained susceptible to entecavir. HBV isolates from lamivudine-refractory
patients failing BARACLUDE therapy were susceptible in vitro to adefovir but retained
resistance to lamivudine.
Lamivudine-resistant strains harboring rtL180M plus rtM204V in combination with amino acid
substitution rtA181C conferred 16- to 122-fold reductions in entecavir phenotypic susceptibility.
Pharmacokinetics
The single- and multiple-dose pharmacokinetics of entecavir were evaluated in healthy subjects
and patients with chronic hepatitis B infection (including liver transplant recipients). Steady-state
Pharmacokinetics of entecavir are summarized in Table 7.
Table 7 - Summary of Entecavir Pharmacokinetic Parameters in Healthy Subjects
Cmax
(ng/mL)
T
1/2
(h)
AUC(TAU)
1
(ng.h/mL)
Clearance
(CLT/F)
(mL/min)
CLR
(mL/min)
Steady-state
mean (0.5 mg)
4.2
130
14.8
572
360
Steady-state
mean (1.0 mg)
8.2
149
26.4
636
471
1
Geometric mean
Absorption
Following oral administration in healthy subjects, entecavir was rapidly absorbed with peak
plasma concentrations occurring between 0.5 and 1.5 hours. Following multiple daily doses
ranging from 0.1 to 1.0 mg, C
max
and area under the concentration-time curve (AUC) at steady
state increased in proportion to dose. Steady state was achieved after 6-10 days of once-daily
administration with approximately 2 fold accumulation. For a 0.5-mg oral dose, C
max
at steady
state was 4.2 ng/mL and trough plasma concentration (C
trough
) was 0.3 ng/mL. For a 1 mg oral
dose, C
max
was 8.2 ng/mL and C
trough
was 0.5 ng/mL.
Effects of food on oral absorption: Oral administration of 0.5 mg of entecavir with a standard
high-fat meal (945 kcal, 54.6 g fat) or a light meal (379 kcal, 8.2 g fat) resulted in a delay in
absorption (1.0-1.5 hours fed vs. 0.75 hours fasted), a decrease in C
max
of 44%-46%, and a
decrease in AUC of 18%-20%. Therefore, BARACLUDE should be administered on an empty
stomach (at least 2 hours after a meal and at least 2 hours before the next meal).

17
Distribution
Based on the pharmacokinetic profile of entecavir after oral dosing, the estimated apparent
volume of distribution is in excess of total body water, suggesting that entecavir is extensively
distributed into tissues. Protein binding to human serum protein in vitro was approximately 13%.
Metabolism
The metabolism of entecavir was evaluated in in vitro and in vivo studies. Entecavir is not a
substrate, inhibitor, or inducer of the cytochrome P450 (CYP450) enzyme system. At
concentrations approximately 10,000 fold higher than those obtained in humans, entecavir
inhibited none of the major human CYP450 enzymes 1A2, 2C9, 2C19, 2D6, 3A4, 2B6, and 2E1.
At concentrations approximately 340 fold higher than those observed in humans, entecavir did
not induce the human CYP450 enzymes 1A2, 2C9, 2C19, 3A4, 3A5, and 2B6. Following
administration of
14
C-entecavir in humans and rats, no oxidative or acetylated metabolites were
observed. Minor amounts of the phase II metabolites glucuronide and sulfate conjugate were
observed.
Excretion
After reaching peak concentration, entecavir plasma concentrations decreased in a bi- exponential
manner with a terminal elimination half-life of approximately 128-149 hours.
The observed drug accumulation index is approximately 2 fold with once-daily dosing, indicating
an effective accumulation half-life of approximately 24 hours.
Entecavir is predominantly eliminated by the kidney with urinary recovery of unchanged drug at
steady state ranging from 62% to 73% of the administered dose. Renal clearance is independent
of dose and ranges from 360 to 471 mL/min suggesting that entecavir undergoes both glomerular
filtration and net tubular secretion (see DRUG INTERACTIONS).
Special Populations and Conditions
Patients Co-Infected with HIV and HBV
Study AI463038 was a randomized, double-blind, placebo-controlled study of BARACLUDE
versus placebo in 68 patients co-infected with HIV and HBV, who experienced recurrence of
HBV viremia while receiving a lamivudine-containing highly active antiretroviral (HAART)
regimen. Patients continued their lamivudine-containing HAART regimen (lamivudine dose 300
mg/day) and were assigned to add either BARACLUDE 1 mg once daily (51 patients) or placebo
(17 patients) for 24 weeks followed by an open-label phase for an additional 24 weeks where all
patients received BARACLUDE. At baseline, patients had a mean serum HBV DNA level by
PCR of 9.13 log 10 copies /mL. Ninety-nine percent of patients were HBeAg-positive at
baseline, with a mean baseline ALT level of 71.5 U/L. Median HIV RNA level remained stable
at approximately 2 log10 copies/mL through 24 weeks of blinded therapy. Virologic and
biochemical endpoints at Week 24 are shown in Table8. There are no data in patients with
HIV/HBV co-infection who have not received prior lamivudine therapy. BARACLUDE has not
been evaluated in HIV/HBV co-infected patients who were not simultaneously receiving
effective HIV treatment. (See WARNINGS AND PRECAUTIONS – Special Populations:
Patients Co-Infected with HIV and HBV)

18
Table 8: Virologic and Biochemical Endpoints at Week 24, Study AI463038
BARACLUDE 1 mg
a
N=51
Placebo
a
N=17
HBV DNA
b
Proportion undetectable (<300 copies/mL)
6%
0
Mean change from baseline (log
10
copies/mL)
(-3.65*)
(+0.11)
ALT normalization (≤ 1 x ULN)
(34%)
c
(8%)
c
a
All patients also received a lamivudine-containing HAART regimen
b
Roche COBAS Amplicor PCR assay (LLOQ = 300 copies/mL)
c
Percentage of patients with abnormal ALT (> 1 x ULN) at baseline who achieved ALT normalization (n=35
for BARACLUDE and n=12 for placebo)
* p < 0.0001
For patients originally assigned to BARACLUDE, at the end of the open-label phase (Week 48),
8% of patients had HBV DNA < 300 copies/mL by PCR, the mean change from baseline HBV
DNA by PCR was -4.20 log
10
copies/mL, and 37% of patients with abnormal ALT at baseline had
ALT normalization (≤ 1 X ULN).
Pediatrics
Pharmacokinetic studies have not been conducted in children.
Geriatrics
The effect of age on the pharmacokinetics of entecavir was evaluated following administration of
a single 1mg oral dose in healthy young (20–40 years old) and elderly (65-83 years old)
volunteers. Entecavir AUC was 29.3% greater in elderly subjects compared to young subjects.
The disparity in exposure between elderly and young subjects was most likely attributable to
differences in renal function. Dosage adjustment of BARACLUDE should be based on the renal
function of the patient, rather than age (see DOSAGE AND ADMINISTRATION: Renal
Impairment).
Gender / race
There are no significant gender/racial differences in entecavir pharmacokinetics.
Hepatic Impairment
No dosage adjustment of BARACLUDE is recommended for patients with hepatic impairment.
The pharmacokinetics of entecavir following a single 1 mg dose were studied in patients (without
chronic hepatitis B infection) with moderate and severe hepatic impairment. The
pharmacokinetics of entecavir were similar between hepatically impaired patients and healthy
control subjects.
Post-liver transplant
The safety and efficacy of BARACLUDE in liver transplant recipients are unknown. However, in
a small pilot study of entecavir use in HBV-infected liver transplant recipients on a stable dose of
cyclosporine A (n=5) or tacrolimus (n=4), entecavir exposure was approximately 2 fold the
exposure in healthy subjects with normal renal function. Altered renal function contributed to the
increase in entecavir exposure in these patients. The potential for pharmacokinetic interactions
between entecavir and cyclosporine A or tacrolimus was not formally evaluated. Renal function
must be carefully monitored both before and during treatment with BARACLUDE in liver
19
transplant recipients who have received or are receiving an immunosuppressant that may affect
renal function, such as cyclosporine or tacrolimus (see DOSAGE AND ADMINISTRATION:
Renal Impairment).
Renal Insufficiency
The pharmacokinetics of entecavir following a single 1 mg dose were studied in patients (without
chronic hepatitis B infection) with selected degrees of renal impairment, including patients whose
renal impairment was managed by hemodialysis or continuous ambulatory peritoneal dialysis
(CAPD). Results are shown in Table 9.
Table 9: Pharmacokinetic Parameters in Subjects with Selected Degrees of Renal
Function
Baseline Creatinine Clearance (mL/min)
Severe
Managed with
Hemodialysis
a
(n = 6)
Severe
Managed
with CAPD
(n = 4)
Unimpaired
> 80
(n = 6)
Mild
> 50≤ 80
(n = 6)
Moderate
30-50
(n = 6)
Severe
< 30
(n = 6)
C
max
(ng/mL)
(CV%)
8.1
(30.7)
10.4
(37.2)
10.5
(22.7)
15.3
(33.8)
15.4
(56.4)
16.6
(29.7)
AUC
(0-T)
(ng·h/mL)
(CV)
27.9
(25.6)
51.5
(22.8)
69.5
(22.7)
145.7
(31.5)
233.9
(28.4)
221.8
(11.6)
CLR (mL/min)
(SD)
383.2
(101.8)
197.9
(78.1)
135.6
(31.6)
40.3
(10.1)
NA NA
CLT/F (mL/min)
(SD)
588.1
(153.7)
309.2
(62.6)
226.3
(60.1)
100.6
(29.1)
50.6
(16.5)
35.7
(19.6)
a
Dosed immediately following hemodialysis
CLR=renal clearance; CLT/F=apparent oral clearance.
Dosage adjustment is recommended for patients with a creatinine clearance <50 mL/min,
including patients on hemodialysis or CAPD. (See DOSAGE AND ADMINISTRATION:
Renal Impairment).
Following a single 1 mg dose of BARACLUDE, hemodialysis removed approximately 13% of
the BARACLUDE dose over 4 hours and CAPD removed approximately 0.3% of the dose over 7
days. BARACLUDE should be administered after hemodialysis.
STORAGE AND STABILITY
BARACLUDE Tablets should be stored in a tightly closed container at 25° C; excursions
permitted between 15-30° C. Keep bottle in the outer carton to protect from light.
DOSAGE FORMS, COMPOSITION AND PACKAGING
BARACLUDE (entecavir) film-coated tablets contains entecavir as the active ingredient.
BARACLUDE film-coated tablets contain the following inactive ingredients: lactose
monohydrate, microcrystalline cellulose, crospovidone, povidone, and magnesium stearate. The
tablet coating contains titanium dioxide, hypromellose, polyethylene glycol 400, polysorbate 80.

20
BARACLUDE Tablets are available in the following strength and configuration of plastic bottles
with child-resistant closures:
Product Strength and
Dosage Form
Description Quantity
0.5 mg film-coated tablet
White to off-white, triangular-shaped tablet, debossed
with “BMS” on one side and “1611" on the other side
30 tablets
21
PART II: SCIENTIFIC INFORMATION
PHARMACEUTICAL INFORMATION
Drug Substance
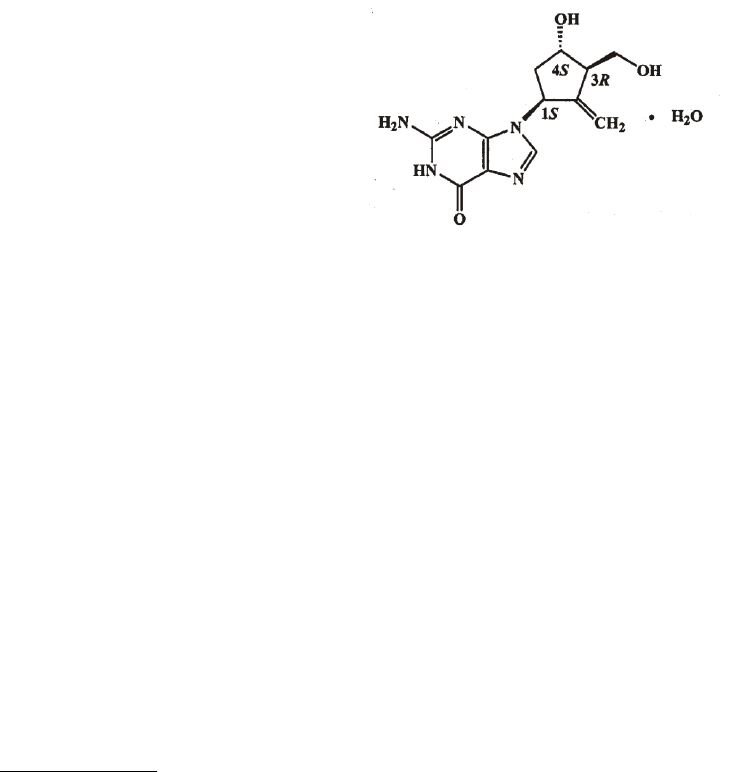
Proper name: Entecavir
Chemical name: 2-amino-1,9-dihydro-9-[(1S,3R,4S)-4-hydroxy-3-(hydroxymethyl)-
2- methylenecyclopentyl]- 6H-purin-6-one, monohydrate.
Molecular formula and molecular mass: C
12
H
15
N
5
O
3
•H
2
O 295.3
Structural formula:
Physicochemical properties: White to off-white powder. It is slightly soluble in water
(2.4 mg/mL), and the pH of the saturated solution in water
is 7.9 at 25° ± 0.5° C.
CLINICAL TRIALS
The safety and efficacy of BARACLUDE were evaluated in three pivotal active-controlled trials
on five continents. These studies included 1633 patients 16 years of age or older with chronic
hepatitis B infection (serum HBsAg-positive for at least 6 months) accompanied by evidence of
viral replication (detectable serum HBV DNA, as measured by the bDNA hybridization or PCR
assay). Subjects had persistently elevated ALT levels ≥ 1.3 times the upper limit of normal
(ULN) and chronic inflammation on liver biopsy compatible with a diagnosis of chronic viral
hepatitis. The safety and efficacy of BARACLUDE were also evaluated in a study of 68 patients
co-infected with HBV and HIV.
Nucleoside-Naive Patients With Compensated Liver Disease, Outcomes at 48 Weeks
HBeAg-positive
Study AI463022 was a multinational, randomized, double-blind study of BARACLUDE 0.5 mg
once daily versus lamivudine 100 mg once daily for a minimum of 52 weeks in 709 (of 715

22
randomized) nucleoside-naive patients with chronic hepatitis B infection and detectable HBeAg.
The mean age of patients was 35 years, 75% were male, 57% were Asian, 40% were Caucasian,
and 13% had previously received interferon-α. At baseline, patients had a mean Knodell
Necroinflammatory Score of 7.8, mean serum HBV DNA as measured by Roche COBAS
Amplicor® PCR assay was 9.66 log
10
copies/mL, and mean serum ALT was 143 U/L. Paired,
adequate liver biopsy samples were available for 89% of patients.
HBeAg-negative (anti-HBe positive/HBV DNA positive)
Study AI463027 was a multinational, randomized, double-blind study of BARACLUDE 0.5 mg
once daily versus lamivudine 100 mg once daily for a minimum of 52 weeks in 638 (of 648
randomized) nucleoside-naive patients with HBeAg-negative (HBeAb-positive) chronic hepatitis
B infection. The mean age of patients was 44 years, 76% were male, 39% were Asian, 58% were
Caucasian, and 13% had previously received interferon-α. At baseline, patients had a mean
Knodell Necroinflammatory Score of 7.8, mean serum HBV DNA as measured by Roche
COBAS Amplicor PCR assay was 7.58 log
10
copies/mL, and mean serum ALT level was 142
U/L. Paired, adequate liver biopsy samples were available for 88% of patients.
In Studies AI463022 and AI463027, BARACLUDE was superior to lamivudine on the primary
efficacy endpoint of Histologic Improvement, defined as ≥ 2-point reduction in Knodell
Necroinflammatory Score with no worsening in Knodell Fibrosis Score at Week 48, and on the
secondary efficacy measures of reduction in viral load and ALT normalization. Histologic
Improvement and change in Ishak Fibrosis Score are shown in Table 10. Biochemical, virologic,
and serologic outcome measures are shown in Table 11.

23
Table 10: Histologic Improvement and Change in Ishak Fibrosis Score at Week 48,
Nucleoside-Naive Patients in Studies AI463022 and AI463027
Study AI463022 (HBeAg-Positive)
Study AI463027 (HBeAg-Negative)
BARACLUDE
0.5 mg
n = 314
a
Lamivudine
100 mg
n = 314
a
Difference
BARACLUDE
Lamivudine
(95% CI)
b
BARACLUDE
0.5 mg
n = 296
a
Lamivudine
100 mg
n = 287
a
Difference
BARACLUDE
Lamivudine
(95% CI)
b
Histologic Improvement (Knodell Scores)
Improvement
c
72% 62%
9.9%
(2.6%, 17.2%)
p < 0.01
70% 61%
9.6%
(2.0%, 17.3%)
p < 0.05
No improvement
21% 24% 19% 26%
Ishak Fibrosis Score
d
Improvement
d
39% 35%
3.2%
(-4.4%, 10.7%)
p= NS
e
36% 38%
-1.8%
(-9.7%, 6.0%)
p= NS
e
No change
46% 40% 41% 34%
Worsening
d
8% 10% 12% 15%
Missing Week
48 biopsy
7% 14% 10% 13%
a
Patients with evaluable baseline histology (baseline Knodell Necroinflammatory Score ≥ 2).
b
In these analyses, missing or inadequate biopsies at Week 48 were classified “no improvement.”
c
≥ 2-point decrease in Knodell Necroinflammatory Score from baseline with no worsening of the
Knodell Fibrosis Score.
d
For Ishak Fibrosis Score, improvement = ≥ 1-point decrease from baseline and worsening = ≥ 1-
point increase from baseline.
e
NS = Not significant.
24
Table 11: Selected Virologic, Biochemical, and Serologic Endpoints at Week 48,
Nucleoside-Naive Patients in Studies AI463022 and AI463027
Study AI463022 (HBeAg-Positive)
Study AI463027 (HBeAg-Negative)
BARACLUDE
0.5 mg
n = 354
Lamivudine
100 mg
n = 355
Difference
BARACLUDE
Lamivudine
(95% CI)
BARACLUDE
0.5 mg
n = 325
Lamivudine
100 mg
n = 313
Difference
BARACLUDE
Lamivudine
(95% CI)
ALT normalization
(
≤ 1.0X ULN)
68 % 60 %
8.4 %
1.3%, 15.4%
p = 0.0202
78 % 71%
6.9 %
a
0.2, 13.7
p = 0.0451
HBV DNA
Mean change from
baseline by
PCR
a
(log
10
copies/mL)
-6.86 -5.39
-1.5
(-1.8 -1.3)
p < 0.0001
-5.04 -4.53
-0.4
(-0.6 -0.3)
p < 0.0001
Proportion
undetectable
(< 300 copies/mL)
by PCR
a, b
67% 36%
30.3%
(23.3%-37.3%)
p < 0.0001
90% 72%
18.3%
(12.3%, 24.2%)
p < 0.0001
<0.7 MEq/mL by
bDNA
c
91% 65%
25.6%
(19.8%, 31.4%)
p < 0.0001
95% 89%
5.9%
(1.8%, 10.1%)
p < 0.01
Loss of HBeAg
22% 20% N/A N/A
HBeAg
seroconversion
21% 18% N/A N/A
a
Roche COBAS Amplicor PCR assay.
b
At Week 24, HBV DNA <300 copies/mL by PCR was observed in 42% of BARACLUDE-treated
patients and 25% of lamivudine-treated patients (p<0.0001) in Study AI463022 and 74% of
BARACLUDE -treated patients and 62% of lamivudine-treated patients (p = 0.0013) in Study AI463027.
c
Quantiplex bDNA assay.
Histologic improvement was independent of baseline levels of HBV DNA or ALT.
Lamivudine-Refractory Patients, Outcomes at 48 Weeks
Study AI463026 was a multinational, randomized, double-blind study of BARACLUDE in 286
(of 293 randomized) HBeAg-positive patients with lamivudine-refractory chronic hepatitis B
infection. Patients receiving lamivudine at study entry either switched to BARACLUDE 1 mg
once daily (with neither a washout nor an overlap period) or continued on lamivudine 100 mg for
a minimum of 52 weeks. The mean age of patients was 39 years, 76% were male, 37% were
Asian, 62% were Caucasian, and 52% had previously received interferon-α. The mean duration
of prior lamivudine therapy was 2.7 years, and 85% had lamivudine resistance mutations at
baseline by an investigational line probe assay. At baseline, patients had a mean Knodell
Necroinflammatory Score of 6.5, mean serum HBV DNA as measured by Roche COBAS
Amplicor PCR assay was 9.36 log
10
copies/mL, and mean serum ALT level was 128 U/L. Paired,
adequate liver biopsy samples were available for 87% of patients.
BARACLUDE was superior to lamivudine on a primary endpoint of Histologic Improvement
(using the Knodell Score at Week 48). These results and change in Ishak Fibrosis Score are
25
shown in Table 12. Table 13 shows selected virologic, biochemical, and serologic endpoints.
Table12: Histologic Improvement and Change in Ishak Fibrosis Score, and Composite
Endpoint at Week 48, Lamivudine-Refractory Patients in Study AI463026
Study AI463026 (HBeAg-Negative)
BARACLUDE
1 mg
n = 124
a
Lamivudine
100 mg
n = 116
a
Difference BARACLUDE
Lamivudine
(97.5% CI)
Histologic Improvement (Knodell Scores)
Improvement
b
55%
28%
27.3%
c
(13.6%, 40.9%)
p < 0.0001
No improvement
34%
57%
Ishak Fibrosis Score
Improvement
d
34%
16%
17.5%
c
(6.8%, 28.2%)
e
p < 0.01
No change
44%
42%
Worsening
d
11%
26%
Inadequate Week 48
biopsy
Missing Week 48 biopsy
2%
10%
1%
15%
a
Patients with evaluable baseline histology (baseline Knodell Necroinflammatory Score ≥ 2).
b
≥ 2-point decrease in Knodell Necroinflammatory Score from baseline with no worsening of the Knodell
Fibrosis Score.
c
In this analysis, missing or inadequate biopsies at Week 48 were classified “no improvement.”
d
For Ishak Fibrosis Score, improvement = ≥1-point decrease from baseline and worsening = ≥ 1-point
increase from baseline.
e
95% confidence interval.
Table 13: Selected Virologic, Biochemical, and Serologic Endpoints at Week 48,
Lamivudine-Refractory Patients in Study AI463026
Study AI463026
BARACLUDE
1 mg
n = 141
Lamivudine
100 mg
n = 145
Difference BARACLUDE
Lamivudine
(95% CI)
ALT normalization (
≤ 1.0 X
ULN)
a
61% 15 %
45.8 %
b
(35.9 %, 55.8 %)
p < 0.0001
HBV DNA
Mean change from baseline
by PCR
a
(log
10
copies/mL)
-5.1 -0.48
-4.4
a
(-4.8, -4.0)
Proportion undetectable
(< 300 copies/mL by PCR
a
)
19% 1%
17.8%
(11.0, 24.5)
p < 0.0001
<0.7 MEq/mL by bDNA
c
66% 6%
60.4%
a
(51.8%, 69.1%)
p < 0.0001
Loss of HBeAg
10%
3%
HBeAg
seroconversion
8% 3%
a
Roche COBAS Amplicor PCR assay.
b
At Week 24, HBV DNA <300copies/mL by PCR was observed in 7% of BARACLUDE-treated patients and no
lamivudine-treated patients (p=0.0011) in Study AI463026
c
Quantiplex bDNA assay.
26
Histologic improvement was independent of baseline levels of HBV DNA or ALT.
Outcomes Beyond 48 Weeks
The optimal duration of therapy with BARACLUDE is unknown. According to protocol-
mandated criteria in the Phase 3 clinical trials, patients discontinued BARACLUDE or
lamivudine treatment after 52 weeks according to a definition of response based on HBV
virologic suppression (<0.7 MEq/mL by bDNA assay) and loss of HBeAg (in HBeAg-positive
patients) or ALT <1.25 X ULN (in HBeAg-negative patients) at Week 48. Patients who achieved
virologic suppression but did not have a serologic response (HBeAg-positive) or did not achieve
ALT <1.25 X ULN (HBeAg-negative) continued blinded dosing through 96 weeks or until
response was achieved. These protocol-specified patient management guidelines are not intended
as guidance for clinical practice.
Nucleoside-naive, outcomes beyond 48 weeks: Cumulative confirmed outcomes through Week 96
for all treated patients in studies of nucleoside-naive patients are shown in Table 14.
Table 14: Outcomes Through 96 Weeks, Nucleoside-Naïve Patients in Studies AI463022
and AI463027 (All Treated)
Study AI463022
(HBeAg-Positive)
Study AI463027
(HBeAg-Negative)
BARACLUDE
0.5 mg
n=354
Lamivudine
100 mg
n=355
BARACLUDE
0.5 mg
N=325
Lamivudine
100 mg
n=313
HBV DNA
a
Proportion undetectable
(< 300 copies/mL)
80%*
39%
94%*
77%
ALT normalization
(≤ 1xULN)
87%*
79%
89%
84%
HBeAg seroconversion
b
31%
26%
NA
NA
HBeAg loss
b
5%
3%
<1%
<1%
a Roche COBAS Amplicor PCR assay (LLOQ = 300 copies/mL).
b through the last observation on or off treatment.
* p<0.01
Among nucleoside-naive HBeAg-positive patients, 243 BARACLUDE-treated and 164
lamivudine-treated patients continued blinded treatment into year 2 (median duration of therapy
was 96 weeks). The proportion of patients with HBV DNA <300 copies/mL by PCR increased
from 64% at Week 48 to 81% at Week 96/EOD [End of Dosing (last observation carried forward)
for patients who discontinued between Weeks 48 and 96] for BARACLUDE-treated patients and
remained stable for lamivudine-treated patients (40% at Week 48 and 39% at Week 96/EOD).
For BARACLUDE-treated patients, ALT normalization (≤1 X ULN) occurred in 66% at Week
48 and 79% at Week 96/EOD. The percentage of lamivudine-treated patients with ALT
normalization was 71% at Week 48 and 68% at Week 96/EOD.
Among nucleoside-naive HBeAg-negative patients, 26 patients continued BARACLUDE
treatment and 28 patients continued lamivudine treatment into year 2 (median duration of therapy
was 96 weeks). The proportion of patients with HBV DNA <300 copies/mL remained stable in
both treatment groups (BARACLUDE 100% at Week 48 and 96% at Week 96/EOD; lamivudine
64% at both Week 48 and Week 96/EOD). No patient in either treatment group had ALT
27
normalization at Week 48, while 27% of BARACLUDE-treated patients and 21% of lamivudine-
treated patients achieved ALT normalization at Week 96/EOD.
Liver biopsy results: Of the 679 BARACLUDE-treated patients in the two nucleoside-naïve
studies, 293 (43%) eligible patients enrolled in a long-term rollover study and continued
BARACLUDE therapy. Patients in the rollover study received BARACLUDE 1 mg once daily.
Sixty-nine of the 293 patients elected to have a repeat liver biopsy after a total treatment duration
of more than 144 weeks (3 years). Fifty-seven patients had both an evaluable baseline and long-
term biopsy, with a median duration of BARACLUDE therapy of 280 weeks (approximately 6
years). Ninety-six percent of these patients had Histologic Improvement as previously defined
(see Table 10, footnote c) and 88% had a ≥ 1-point decrease in Ishak fibrosis score. Of the 43
patients with a baseline Ishak fibrosis score of ≥ 2, 58% had a ≥ 2-point decrease. At the time of
the long-term biopsy, 57 (100%) of patients had HBV DNA < 300 copies/mL and 49 (86%) had
serum ALT ≤ 1 X ULN.
Lamivudine-refractory, outcomes beyond 48 weeks: Cumulative confirmed outcomes through
Week 96 for all treated lamivudine-refractory patients are shown in Table 15.
Table 15: Outcomes Through 96 Weeks, Lamivudine Refractory Patients in Study
AI463026 (All Treated)
BARACLUDE
1 mg
n= 141
LAMIVUDINE
100 mg
n=145
HBV DNA
a
Proportion undetectable (<300 copies/mL)
30%*
<1%
ALT normalization (≤ 1 x ULN)
85%*
29%
HBeAg seroconversion
b
17%*
6%
a Roche COBAS Amplicor PCR assay (LLOQ = 300 copies/mL).
b through the last observation on or off treatment.
*p<0.01
Among lamivudine-refractory patients in Study AI463026, 77 BARACLUDE-treated patients
continued dosing into year 2 (median duration of therapy was 96 weeks). The proportion of
patients with HBV DNA <300 copies/mL increased from 21% at Week 48 to 40% at Week
96/EOD. The proportion of patients with ALT normalization increased from 65% at Week 48 to
81% at Week 96/EOD.
Post-Treatment Follow-up
For the 31% of nucleoside-naive, HBeAg-positive BARACLUDE-treated patients who met
response criteria (virologic suppression by bDNA assay and loss of HBeAg) and discontinued
therapy, response was sustained throughout the 24-week post-treatment follow-up period in 75%.
For the 88% of nucleoside-naive, HBeAg-negative BARACLUDE-treated patients who met
response criteria (virologic suppression by bDNA assay and ALT <1.25 X ULN), response was
sustained throughout the 24-week post-treatment follow-up period in 46%. Of the 22 (16%)
lamivudine-refractory patients who met response criteria (virologic response on bDNA assay and
loss of HBeAg) while receiving BARACLUDE, response was sustained throughout the 24-week
post-treatment follow-up period in 11 (50%).
28
Data from Long-term Observational Study
Study AI463080 was a randomized, global, observational, open-label Phase 4 study to assess
long-term risks and benefits of BARACLUDE (0.5 mg/day or 1 mg/day) treatment as compared
to other standard of care hepatitis B virus nucleos(t)ide analogues in subjects with chronic HBV
(CHB) infection.
A total of 12,378 patients were treated with Baraclude (n=6,216) or other HBV nucleos(t)ide
(non-entecavir (ETV) (n=6,162). The patients were evaluated at baseline and subsequently every
6 months for up to 10 years. The principal clinical outcome events assessed during the study were
overall malignant neoplasms, liver-related HBV disease progression, non-HCC malignant
neoplasms, HCC, and deaths. The study showed that Baraclude was not significantly associated
with an increased risk of malignant neoplasms compared to other standard of care HBV
nucleos(t)ides, as assessed by either the composite endpoint of overall malignant neoplasms or
the individual endpoint of non-HCC malignant neoplasm. The most commonly reported
malignancy was HCC followed by gastrointestinal malignancies in both the Baraclude and non-
ETV groups. The data also showed that long-term Baraclude use was not associated with a lower
occurrence of HBV disease progression or a lower rate of death overall compared to other HBV
nucleo(t)ides. The principal clinical outcome event assessments are shown in Table 16:
Table 16: Principal Analyses of Time to Adjudicated Events - Randomized Treated
Subjects
Number of Subjects with
Events
Endpoint
a
ETV
N=6,216
Non-ETV
N=6,162
Hazard Ratio
[ETV:Non-ETV] (CI)
b
P-value
c
Primary Endpoints
Overall malignant neoplasm 331 337 0.93 (0.800, 1.084) 0.3553
Liver-related HBV disease progression
350
375
0.89 (0.769, 1.030)
0.1182
Death
238
264
0.85 (0.713, 1.012)
0.0676
Secondary Endpoints
Non-HCC malignant neoplasm
95
81
1.10 (0.817, 1.478)
HCC
240
d
263 0.87 (0.727, 1.032)
a
Overall malignant neoplasm is a composite event of HCC or non-HCC malignant neoplasm. Liver-related HBV
disease progression is a composite event of liver-related death, HCC, or non-HCC HBV disease progression.
b
95.03% CI for overall malignant neoplasm, death, and liver-related HBV disease progression; 95% CI for non-
HCC malignant neoplasm, HCC, liver-related death, and non-HCC HBV disease progression.
c
P-values are provided to the COEs that are primary endpoints per protocol specification
d
One subject had a pre-treatment HCC event and was excluded from the analysis.
CI = confidence interval; N = total number of subjects.
Limitations of the study included population changes over the long-term follow-up period and
more frequent post-randomization treatment changes in the non-ETV group. In addition, the
29
study was underpowered to demonstrate a difference in the non-HCC malignancy rate because of
the lower than expected background rate.
Special Populations
Patients Co-Infected with HIV and HBV
Study AI463038 was a randomized, double-blind, placebo-controlled study of BARACLUDE
versus placebo in 68 patients co-infected with HIV and HBV who experienced recurrence of
HBV viremia while receiving a lamivudine-containing highly active antiretroviral (HAART)
regimen. Patients continued their lamivudine-containing HAART regimen (lamivudine dose 300
mg/day) and were assigned to add either BARACLUDE 1 mg once daily (51 patients) or placebo
(17 patients) for 24 weeks followed by an open-label phase for an additional 24 weeks where all
patients received BARACLUDE. At baseline, patients had a mean serum HBV DNA level by
PCR of 9.13 log
10
copies/mL. Ninety-nine percent of patients were HBeAg-positive at
baseline, with a mean baseline ALT level of 71.5 U/L. Median HIV RNA level remained stable at
approximately 2 log
10
copies/mL through 24 weeks of blinded therapy. Virologic and
biochemical endpoints at Week 24 are shown in Table16. There are no data in patients with
HIV/HBV co-infection who have not received prior lamivudine therapy. BARACLUDE has not
been evaluated in HIV/HBV co-infected patients who were not simultaneously receiving
effective HIV treatment. (See WARNINGS AND PRECAUTIONS – Special Populations:
Patients Co-infected with HIV and HBV).
Table 17: Virologic and Biochemical Endpoints at Week 24, Study AI463038
BARACLUDE 1 mg
a
n = 51
Placebo
a
n = 17
HBV DNA
b
Proportion undetectable (< 300 copies/mL)
6%
0
Mean change from baseline (log
10
copies/mL)
(-3.65*)
(+0.11)
ALT normalization (≤ 1 x ULN)
(34%)
c
(8%)
c
a
All patients also received a lamivudine-containing HAART regimen
b
Roche COBAS Amplicor PCR assay (LLOQ = 300 copies/mL)
c
Percentage of patients with abnormal ALT (> 1 x ULN) at baseline who achieved ALT normalization (n=35 for
BARACLUDE and n=12 for placebo)
* p < 0.0001
For patients originally assigned to BARACLUDE, at the end of the open-label phase (Week 48),
8% of patients had HBV DNA < 300 copies/mL by PCR, the mean change from baseline HBV
DNA by PCR was -4.20 log10 copies/mL, and 37% of patients with abnormal ALT at baseline
had ALT normalization (≤ 1% ULN).
DETAILED PHARMACOLOGY
Mechanism of Action
Entecavir is a guanosine nucleoside analogue that is efficiently phosphorylated to the active
triphosphate form and exhibits selective activity against HBV polymerase. Entecavir triphosphate
competes with the natural substrate deoxyguanosine triphosphate, and inhibits all three functional
30
activities of the HBV polymerase (reverse transcriptase, rt): (1) base priming, (2) reverse
transcription of the negative strand from the pregenomic messenger RNA, and (3) synthesis of
the positive strand of HBV DNA. Entecavir triphosphate has an inhibition constant (Ki) for HBV
DNA polymerase of 0.0012 μM and is a weak inhibitor of cellular DNA polymerases α, ß, and δ
and mitochondrial DNA polymerase γ with Ki values ranging from 18 to >160 μM.
Antiviral Activity
Entecavir inhibited HBV DNA synthesis (50% reduction, EC
50
) at a concentration of 0.004 µM
in human HepG2 cells transfected with wild-type HBV. The median EC
50
value for entecavir
against lamivudine-resistant HBV (rtL180M, rtM204V) was 0.026 µM (range 0.010-0.059 µM).
A comprehensive analysis of the inhibitory activity of entecavir against a panel of laboratory and
clinical HIV-1 isolates using a variety of cells and assay conditions yielded EC
50
values ranging
from 0.026 to >10 µM: the lower EC
50
values were observed when decreased levels of virus were
used in the assay. In cell culture, entecavir selected for an M184I substitution in HIV reverse
transcriptase at micromolar concentrations, confirming inhibitory pressure at high entecavir
concentrations. HIV variants containing the M184I substitution showed loss of susceptibility to
entecavir.
Daily or weekly entecavir treatment significantly reduced viral DNA levels (4 to 8 log10) in two
relevant animal models, woodchucks chronically infected with woodchuck hepatitis virus (WHV)
and ducks infected with duck HBV. Long-term studies in woodchucks demonstrated that oral
weekly dosing of 0.5 mg/kg entecavir (similar exposure to the 1 mg human dose) maintained
viral DNA levels at undetectable levels (<200 copies/mL by PCR) for up to 3 years in 3 of 5
woodchucks. No entecavir resistance changes were detected in the HBV polymerase in any of the
treated animals for up to 3 years of treatment.
TOXICOLOGY
Acute Toxicity (Table 1)
Single-dose oral toxicity studies with entecavir were conducted in mice and rats at doses ranging
from 40 to 5000 mg/kg. In mice, no drug-related changes were noted at 40 mg/kg. Body-weight
losses were noted at ≥ 200 mg/kg. At ≥1000 mg/kg, signs of overt toxicity and deaths were
observed. In rats, no drug-related changes were observed at doses of 40 or 200 mg/kg. Deaths
occurred at ≥ 1000 mg/kg.
Repeat-Dose Toxicity (Table 2)
Repeat-dose studies utilizing once daily oral dosing were conducted in mice, rats, dogs, and
monkeys. Pivotal studies included two 6-month oral studies each in mice and rats to assess
chronic toxicity and to aid in the selection of doses for oral carcinogenicity studies; two 3-month
studies in dogs to assess toxicity and reversibility of drug-related changes, and a 1-year toxicity
study in monkeys that included a 3-month interim evaluation.

31
In dogs, species-specific, reversible CNS inflammation was observed at doses that achieved ≥ 51
times the exposure to entecavir in humans at 1 mg. The species-specificity, reversibility, and
high exposure multiples at which the CNS inflammation was observed suggest that this finding is
not relevant to human safety. Other target organs in repeat-dose studies in animals were the
kidneys, liver, lungs, skeletal muscle and testis; the changes in these organs were considered
unlikely to be relevant to human safety because they were either species-specific, associated with
high exposure multiples relative to humans, and/or, in clinical trials with entecavir, they were not
target tissues. With regard to target-organ toxicity in general, the results of a 1-year study in
monkeys were most compelling because no target organ toxicity was evident at ≥ 136 times the
exposure to entecavir in humans at 1 mg.
Reproductive Toxicology (Table 3)
Reproductive toxicology studies were conducted with entecavir to assess potential effects on
embryonic and fetal development in rats and rabbits and on growth, development, and
reproductive performance of progeny in rats.
In rats and rabbits, no embryotoxicity or maternal toxicity was observed at 28 and 212 times,
respectively, the exposure to entecavir at 1 mg in humans. In rats, maternal toxicity, embryo-fetal
toxicity (resorptions), and associated decreases in live-litter size occurred at 180 times the
exposure in humans at 1 mg. Additional findings in rat fetuses at 3100 times the exposure in
humans at 1 mg included lower body weights, tail and vertebral malformations, reduced
ossification (vertebrae, sternebrae, and phalanges), and extra lumbar vertebrae and ribs. In
rabbits, embryo-fetal toxicity (resorptions), reduced ossification (hyoid), and an increased
incidence of 13th rib were observed at 883 times the exposure in humans at 1 mg. In a peri-
postnatal study in rats, no adverse effects on offspring were seen at > 94 times the exposure in
humans at 1 mg.
Carcinogenesis, Mutagenesis, Impairment of Fertility (Tables 4 & 5)
Long-term oral carcinogenicity studies of entecavir in mice and rats were carried out at exposures
up to approximately 42 times (mice) and 35 times (rats) those observed in humans at 1 mg.
MICE: Lung adenomas were increased in males and females at exposures 3 and 40 times those
in humans at 1 mg, respectively. Lung carcinomas in both male and female mice were increased
at exposures approximately 40 times those in humans at 1 mg. Lung tumor development was
preceded by pneumocyte proliferation in the lung, which was not observed in rats, dogs, or
monkeys administered entecavir, indicating that a key event in lung tumor development observed
in mice likely was species specific. At the highest dose level tested (equivalent to approximately
40 times the exposure in humans at 1 mg) hepatocellular carcinomas and the combined incidence
of adenomas and carcinomas in male mice and vascular tumors (hemangiomas of ovaries and
uterus and hemangiomas/hemangiosarcomas of spleen) in female mice were significantly
increased.
The NOEL for neoplasia was 0.004 mg/kg/day for males (equivalent to 1-times the exposure in
humans at 1 mg), based on pulmonary adenomas; for all other tumors in male and female mice
the NOEL was 0.4 mg/kg (equivalent to 14 and 11 times the exposure in humans at 1 mg for

32
male and female mice, respectively). At tumorigenic doses, systemic exposures were 3-times
(pulmonary tumors in male mice) and approximately 40-times (all other tumors) that in humans
at 1 mg daily.
RATS: In female rats, hepatocellular adenomas and the combined incidence of adenomas and
carcinomas were significantly increased at the highest dose level, equivalent to 24-times the
exposure in humans at 1 mg. Brain microglial tumors were significantly increased in both male
and female rats at the highest dose, equivalent to 35 and 24 times exposure in humans at 1 mg
respectively. Skin fibromas in female rats were significantly increased at the 0.4 (high) and 2.6
mg/kg/day (highest) doses, equivalent to 4 and 24 times exposure in humans at 1 mg respectively.
The NOEL for neoplasia was 0.2 mg/kg/day for males (equivalent to 5 times the exposure in
humans at 1 mg) and for females 0.06 mg/kg/day based on skin fibromas (equivalent to < 1 times
the exposure in humans at 1 mg) or 0.4 mg/kg/day (all other tumors, equivalent to 4 times the
exposure in humans at 1 mg). At tumorigenic doses, systemic exposures were 35 and 4/24 times
that in humans at 1 mg in male and female rats, respectively.
It is not known how predictive the results of rodent carcinogenicity studies may be for humans.
(see CLINICAL TRIALS, Data from Long-term Observational Study).
Entecavir was clastogenic to human lymphocyte cultures and in mouse lymphoma cells in vitro.
Entecavir was not mutagenic in the Ames bacterial reverse mutation assay, a mammalian-cell
gene mutation assay, and a transformation assay with Syrian hamster embryo cells. Entecavir was
also negative in an oral micronucleus study and an oral DNA repair study in rats. In reproductive
toxicology studies in which rats were administered entecavir at up to 30 mg/kg for up to 4 weeks,
no evidence of impaired fertility was seen in males or females at systemic exposures >90 times
those in humans at 1 mg. In rodent and dog toxicology studies, seminiferous tubular degeneration
was observed at ≥ 35 times the exposure in humans at 1 mg. No testicular changes were evident
in monkeys administered entecavir for 1 year at 167 times the exposure in humans at 1 mg.
33
Table 1 - ACUTE TOXICITY
Species/
Strain
N/Dose/
Sex
Dose
mg/kg/day
Route of
Administration
Observed
Maximum
Non-Lethal
Dose (mg/kg)
Approximate
Lethal Dose
(mg/kg)
Findings
Mouse/CD-1
M5 F5
0, 40, 200,
1000, &
5000
Oral, gavage
200
≥ 1000
40 mg/kg: No drug-related findings.
≥
200 mg/kg: Transient body-weight losses.
≥
1000 mg/kg: One male at 1000 mg/kg and 4
males and all females at 5000 mg/kg died. In the
testis, moderate degeneration of seminiferous
tubular epithelium. In the spleen, mild to moderate
lymphoid depletion.
Rat/SD
M5
0, 40, 200,
1000, &
5000
Oral, gavage
200
≥ 1000
≤ 200 mg/kg: No drug-related findings.
≥
1000 mg/kg: One rat at 1000 mg/kg and all rats
at 5000 mg/kg died.
In rats that died, red
discoloration of small intestine associated with
hemorrhage, and necrosis of duodenum and
jejunum.

34
Table 2 – REPEAT-DOSE TOXICITY
Species/Strain
N/Sex
Dose mg/kg/day
Route of
Administration
Duration of
Dosing
NOAEL
(mg/kg)
Findings
MOUSE
Mouse/CD-1
Four groups of
10M 10F each
0, 0.2, 1, & 5
Oral, gavage
6 months
< 0.2
≥ 0.2 mg/kg: In the liver, minimal to moderate
centrilobular degeneration.
≥ 1 mg/kg: In the lung, minim
al to mild alveolar
histiocytosis. In skeletal muscle, minimal to mild
myopathy.
5 mg/kg
: Lower body weight (males); minimal to
moderate decreases i
n total leukocyte and lymphocyte
counts; mild to moderate increases in serum ALT
and/or AST; increased kidney and decreased testes
weights in males; and minimal to mild centrilobular
hepatocellular hypertrophy in the liver.
Mouse/CD-1
Three groups of
10 M 10F each
0, 10, & 20
Oral, gavage
6 months
< 10
≥ 10 mg/kg: Mortality; lower body weights, minimal to
mild decreases in serum total protein, albumin, and
globulins; decreased weights of the testes and
prostate/seminal vesicles;
increased severity of
nephropathy in the kidneys; minimal to moderate
degeneration and hypertrophy in the centrilobular
region of the liver;
bronchioloalveolar hyperplasia
and/or adenomas, and minimal to moderate alveolar
histiocytosis in the lungs; minimal to moderate skeletal
mus
cle myopathy; and minimal to moderate
seminiferous-tubular degeneration in the testes.
20 mg/kg
: Mild decreases in total leukocyte and
lymphocyte counts in males; and minimal to mild
decreases in serum total protein, albumin, and
globulins.
RAT
Rat/SD
Four groups of
20M 20F each
0, 0.02, 0.08, &
0.3
Oral, gavage
6 months
< 0.02
≥ 0.02 mg/kg: Minimal centrilobular degeneration in
the liver.
Rat/SD
Four groups of
20M 20F each
0, 0.6, 3, & 15
Oral, gavage
6 months
0.6
≥ 0.6 mg/kg: Minimal to mild decreases in serum total
protein, albumin, and globulins in males; minimal to
mild centrilobular degeneration in the liver (associated
with enlarged hepatocellular mitochondria in males;
clearly different than the hepatocellular
megamitochondria reported for another antiviral

35
Species/Strain
N/Sex
Dose mg/kg/day
Route of
Administration
Duration of
Dosing
NOAEL
(mg/kg)
Findings
nucleoside analog and considered secondary to the
hepatocellular degeneration); and minimal to mild
skeletal-muscle myopathy.
≥ 3 mg/kg
: Minimal to mild increases in serum urea
nitrogen and cholesterol
and minimal to moderate
increases in serum AST in males.
15 mg/kg: Lower body weight in males; mild increase
in total leukocyte count in males; minimal to mild
increases in prothrombin time;
minimal to mild
increases in serum urea nitrogen, cholesterol, and
sodium in females; minimal to mild increases in serum
ALT
and chloride; mild increases in water consumption
and urine volume and a mild decrease in urine specific
gravity in males; and decreased testes weight and size.
DOG
Dog/Beagle
Four groups of
3M 3F each
0, 0.3, 3, & 30/15
(due to overt
toxicity at 30
mg/kg, the high
dose was reduced
to 15 mg/kg on
dose day 29.)
Oral, capsule
3 months
< 0.3
≥ 0.3 mg/kg: Decreased weights of the testes and
prostrate; and minimal to moderate inflammation in the
brain.
≥ 3 mg/kg: Decreased weights of the ovaries; minimal
to mild inflammation in the spinal cord; inflammation
and depletion of zymogen granules in the pancreas;
degeneration of seminiferous tubules in the testes; and
atrophy in the prostate.
30/15 mg/kg: Three dogs sacrificed in moribund
condition after approximately 1 month of dosing. In
surviving high-
dose dogs: clinical signs of toxicity,
clinical laboratory changes including mild to moderate
decreases in RBC/WBC parameters and platelet counts;
a mild increase in serum gamma glutamyl transferase;
increased myeloid/erythroid ratio and a moderately
reduced number of megakaryocytes in bone-marrow
smears; and a
moderate increase in urine specific
gravity. In sacrificed and surviving dogs:
myeloid/erythroid depletion in the bone marrow; and
lymphoid depletion in lymph nodes.
Dog/Beagle
Control 4M 4F;
0.1 mg/kg 3M 3F;
15 mg/kg 6M 6F.
0, 0.1, & 15
Oral, capsule
3 months
0.1
0.1 mg/kg: No drug-related changes.
15 mg/kg: Changes generally were consistent with

36
Species/Strain
N/Sex
Dose mg/kg/day
Route of
Administration
Duration of
Dosing
NOAEL
(mg/kg)
Findings
2M 2 F control
and 3M 3F at 15
mg/kg evaluated
after 3-month
postdose period.
those at 30/15 mg/kg in the initial 3-month study, but
target organs were limited to CNS, pancreas, and testes;
all changes were reversible or showed evidence of
reversibility (testes weight).
MONKEY
Monkey/Cynomolgus
Four groups of
6M 6F each; 2M
2F used for
interim evaluation
after 3 months of
dosing
0, 0.4, 4, & 40
Oral, gavage
1 year
40
0.4 and 4 mg/kg: No drug-related changes.
40 mg/kg: Minimal increases in serum urea nitrogen
and potassium.

37
Table 3 – REPRODUCTION AND TERATOLOGY
Study Type Species/Strain
N/ Sex
Dose mg/kg/day
(multiple of human
exposure
Duration of
dosing
Route of
Administration
Findings
Oral Study of Fertility and
Early Embryonic
Development-Treated Female
Rats
Rats/SD
Four groups
of 25 F each
0, 0.3, 3, & 30
2 Weeks
premating
through
Gestation Day 7
Oral, gavage
≥ 0.3 mg/kg: No drug-related changes.
Oral Study of Fertility and
Early Embryonic
Development-Treated Male
Rats
Rats/SD
Four groups of
25 M each
0, 0.1, 1, & 10
4 Weeks
premating until
scheduled
euthanasia (33
to 42 daily
doses)
Oral, gavage
0.1 and 1 mg/kg: No drug-related changes.
10 mg/kg: Decreased body weight and body-weight
gain.
Oral Study of Embryo-Fetal
Development in Rats
Rats/SD
Four groups of
22 F each
0, 2, 20, & 200
Gestation
Days 6-15
Oral, gavage
0.2 mg/kg: No drug-related changes.
≥ 20 mg/kg: In the dams: lower body weights and body-
weight gains. In the fetuses: increases in embryo-fetal
death (resorptions) with associated decreases in live-
litter sizes.
200 mg/kg: In the dams: 1 death, decreased food
consumption, and increased incidence of reduced/absent
feces. In the fetuses: lower body weight; tail and
vertebrae malformations; delays in ossification of the
vertebrae, sternebrae and phalanges; and increases in
the number of lumbar vertebrae and ribs.
Oral Study of Embryo-Fetal
Development in Rabbits
Rabbit/NZW
Four groups of
20 F each
0, 1, 4, & 16
Gestation
Days 6-18
Oral, gavage
1 and 4 mg/kg: No drug-related changes.
16 mg/kg: In the fetuses: increases in embryo-fetal death
(resorptions) with associated decreases in live-litter sizes;
developmental delays in ossification of the hyoid; and an
increased incidence of 13th rib.
Oral Study of Pre- and
Postnatal Development in Rats
Rats/SD
Four groups of
25 F each
0, 0.3, 3, & 30
Gestation Day 6
through Day 20
of Lactation
Oral, gavage
0.3 and 3 mg/kg: No drug-related changes.
30 mg/kg: In the dams: reduced body-weight gain.
38
Table 4 – CARCINOGENICITY
Species/Strain
N/ Sex
Dose
mg/kg/day
Route of
Administration
Duration of
Dosing
Findings
Carcinogenicity
Study in Mice
Mouse/CD-1
Four groups
of
60M, 60F
each
0, 0, 0.004,
0.04, 0.4, & 4
Oral, gavage
24 months
0.004 mg/kg: No drug-related changes.
≥ 0.04 mg/kg: Increased incidences of bro
nchiolo/alveolar adenoma in the lungs
in males.
≥ 0.4 mg/kg
: Increased mortality; hyperplasia of alveolar epithelium,
leukocytosis/interstitial inflammation, and infiltration of the alveolar spaces by
alveolar macrophages in the lungs.
4 mg/kg: Lower body weights and body-weight gains; increased incidences of
focal hyperplasia of bronchiolar epithelium and alveolar fibrosis in the lungs and
hematocyts, thrombi and ectasia in the ovaries; and increased tumor incidences
including: bronchiolo/alveolar carcinoma in the lung
, bronchiolo/alveolar
adenoma (females) in the lungs, hepatocellular carcinoma (males) in the liver,
and vascular tumors (females).
Carcinogenicity
Study in Rats
Rat/SD
Six groups
of 60M, 60F
each
0, 0, 0.003,
0.02, & 0.2,
and 1.4 (M); 0,
0, 0.01, 0.06,
0.4, & 2.6 (F )
Oral, gavage
24 months
0.003 mg/kg (males) and 0.01 and 0.06 mg/kg (females): No drug-related
changes.
≥ 0.2 mg/kg (males): Increased incidences of focal hyperplasia in the acinar
(exocrine) pancreas and hepatocellular alterations in the liver.
1.4 mg/kg (males): Decreased body weight
; increased incidences of
hepatocellular vacuolation, testicular degeneration, and chronic progressive
nephropathy; and increased tumor incidences including malignant glioma in the
brain.
≥0.4 mg/kg (females): Increased incidences of hepatocellular alterations.
2.6 mg/kg (females): Increased incidence of hepatocellular vacuolation; and
increased tumor incidences including hepatocellular adenoma carcinoma
malignant glioma in the brain, and skin fibroma.
39
Table 5 – MUTAGENICITY
Test/Test System
Sex
Concentration/Dose
Route of
Administration
Duration of
Dosing
Effects
IN VITRO
Ames/S. typhimurium
and E. Coli
NA
312.5 to 5000 ng/plate,
both with and without
metabolic activation
NA
48 hr
Not mutagenic
Gene Mutation/CHO
cells-HGPRT locus
NA
50 to 1000 µg/mL
without metabolic
activation
NA
4 hr
Not mutagenic when tested
to cytotoxic concentrations
Cytogenetics/Primary
Human Lymphocytes
NA
2.5 to 20 µg/mL
(without metabolic
activation) and 2.5 to
200 µg/mL with
metabolic activation
NA
24 hr without
metabolic
activation and 5 hr
with metabolic
activation
Cytotoxicity at ≥ 5 µg/mL
(without metabolic
activation) and at 200
µg/mL (with metabolic
activation. Increased
chromosome aberrations at
≥ 10 µg/mL (without
metabolic activation) and at
≥ 50 µg/mL with metabolic
activation.
Cell
transformation/Syrian
hamster embryo cells
NA
0.125 to 2.0 µg/mL
NA
7 days
No increase in
morphologically
transformed cells when
tested to cytotoxic
concentrations
IN VIVO
Micronucleus/Rat
Male
2 to 2000 mg/kg daily
Oral, gavage
3 days
Not genotoxic
DNA Repair/Rat
Male
2 to 2000 mg/kg
Oral, gavage
Single Dose
Not genotoxic
Page 40 of 43
IMPORTANT: PLEASE READ
PART III: CONSUMER INFORMATION
Pr
BARACLUDE*
(entecavir)
This leaflet is Part III of a three-part “Product
Monogaph” published when BARACLUDE was
approved for sale in Canada and is designed
specifically for Consumers. This leaflet is a
summary and will not tell you everything about
BARACLUDE. Contact your doctor or
pharmacist if you have any questions about the
drug.
ABOUT THIS MEDICATION
What the medication is used for
BARACLUDE is a prescription medicine used for
chronic infection with hepatitis B virus (HBV) in
adults who also have active liver damage.
What it does
BARACLUDE may lower the amount of HBV in
the body
BARACLUDE may lower the ability of HBV to
multiply and infect new liver cells
BARACLUDE may reduce the damage to the liver by
HBV
BARACLUDE will not cure HBV infection.
It is important to stay under your healthcare provider’s
care while taking BARACLUDE. Your healthcare
provider will test the level of the hepatitis B virus in
your blood regularly.
When it should not be used
Do not take BARACLUDE if you are allergic to any
of its ingredients. The active ingredient in
BARACLUDE is entecavir. See “What the
nonmedicinal ingredients are” for a complete list of
ingredients in BARACLUDE.
Tell your healthcare provider if you think you have
had an allergic reaction to any of these ingredients.
BARACLUDE has not been studied in children and is
not recommended for anyone less than 16 years old.
What the medicinal ingredient is:
Entecavir
What the nonmedicinal ingredients are:
BARACLUDE tablets: lactose monohydrate,
microcrystalline cellulose, crospovidone, povidone,
magnesium stearate, titanium dioxide, hypromellose,
polyethylene glycol 400, polysorbate 80.
What dosage forms it comes in:
BARACLUDE film-coated tablets
Does BARACLUDE lower the risk of passing HBV
to others?
BARACLUDE does not stop you from spreading
HBV to others by sex, sharing needles, or being
exposed to your blood. Talk to your healthcare
provider about safe sexual practices that protect your
partner. Never share needles. Do not share personal
items that can have blood or body fluids on them, like
toothbrushes or razor blades. A shot (vaccine) is
available to protect people at risk from becoming
infected with HBV.
WARNINGS AND PRECAUTIONS
Serious Warnings and Precautions
Severe worsening of hepatitis (liver inflammation) has
occurred in patients who have stopped taking anti-
hepatitis B therapy (including BARACLUDE). Your
doctor will monitor your condition in this case and
may resume therapy.
Lactic acidosis (increase in acid level of blood) and
severe hepatomegaly with steatosis (enlarged fatty
liver), including fatal cases have been reported in
patients using nucleoside analogue medicines,
including BARACLUDE, either alone or in
combination. Reports of lactic acidosis with
BARACLUDE often involved patients who were
seriously ill due to their liver disease or other medical
condition. Lactic acidosis is a medical emergency and
must be treated in the hospital. Call your healthcare
provider right away if you get any of the signs of
lactic acidosis (see table Serious Side Effects and
What to do About them).

Page 41 of 43
Your hepatitis B infection may get worse or become
very serious if you stop BARACLUDE.
take BARACLUDE exactly as prescribed
do not run out of BARACLUDE
do not stop BARACLUDE without talking to
your healthcare provider
Your healthcare provider will need to monitor your
health and do regular blood tests to check your liver if
you stop BARACLUDE. Tell your healthcare
provider right away about any new or unusual
symptoms that you notice after you stop taking
BARACLUDE.
If you have or get HIV (human immunodeficiency
virus) infection be sure to discuss your treatment with
your doctor. If you are taking BARACLUDE to treat
chronic hepatitis B and are not taking medicines for
your HIV at the same time, some HIV treatments that
you take in the future may be less likely to work. You
are advised to get an HIV test before you start taking
BARACLUDE and anytime after that when there is a
chance you were exposed to HIV. BARACLUDE
will not help your HIV infection.
BEFORE you use BARACLUDE talk to your
healthcare provider about all of your medical
conditions, including if you:
have had a liver transplant.
have kidney problems. Your doctor may need to
adjust your BARACLUDE dose or dose schedule.
are pregnant or planning to become pregnant. It is
not known if BARACLUDE is safe to use during
pregnancy. It is not known whether
BARACLUDE helps prevent a pregnant mother
from passing HBV to her baby. You and your
healthcare provider will need to decide if
BARACLUDE is right for you. If you use
BARACLUDE while you are pregnant, talk to
your healthcare provider about the BARACLUDE
Pregnancy Registry.
are breast-feeding. It is not known if
BARACLUDE can pass into your breast milk or
if it can harm your baby. Do not breast-feed if
you are taking BARACLUDE.
are lactose intolerant. BARACLUDE tablets
contain lactose. If you have been told that you
have intolerance to some sugars, contact your
doctor before taking this medicine.
Tell your healthcare provider about all the medicines
you take including prescription and non-prescription
medicines, vitamins, and herbal supplements.
BARACLUDE may interact with other medicines that
leave the body through the kidneys.
Know the medicines you take. Keep a list of your
medicines with you to show your healthcare provider
and pharmacist.
INTERACTIONS WITH THIS MEDICATION
BARACLUDE may interact with other medicines that
leave the body through the kidneys.
PROPER USE OF THIS MEDICATION
Take BARACLUDE exactly as prescribed. Your
healthcare provider will tell you how much
BARACLUDE to take. Your dose will depend on
whether you have been treated for HBV infection
before and what medicine you took.
Usual dose
The usual dose of BARACLUDE Tablets in adults
and children over 16 years of age is either one or two
0.5 mg tablets once daily by mouth. Your dose may
be lower or you may take BARACLUDE less often
than once a day, if you have kidney problems.
Take BARACLUDE once a day on an empty
stomach to help it work better. Empty stomach
means at least 2 hours after a meal and at least 2
hours before the next meal. To help you
remember to take your BARACLUDE, try to take
it at the same time each day.
Do not change your dose or stop taking
BARACLUDE without talking to your healthcare
provider. Your hepatitis B symptoms may get
worse or become serious if you stop taking
BARACLUDE. After you stop taking
BARACLUDE, it is important to stay under your
healthcare provider’s care. Your healthcare
provider will need to do regular blood tests to
check your liver.
When your supply of BARACLUDE starts to run
low, get more from your healthcare provider or
pharmacy. Do not run out of BARACLUDE.
Overdose
In case of drug overdosage, contact a
healthcare practitioner (e.g. doctor) hospital
emergency department or regional poison
control centre, even if there are no symptoms.
Page 42 of 43
Missed Dose
If you forget to take BARACLUDE, take it as soon as
you remember and then take your next dose at its
regular time. If it is almost time for your next dose,
skip the missed dose. Do not take two doses at the
same time. Call your healthcare provider or
pharmacist if you are not sure what to do.
SIDE EFFECTS AND WHAT TO DO ABOUT
THEM
The most common side effects of BARACLUDE are
headache, tiredness, dizziness, and nausea. Rash has
also been reported. Less common side effects include
diarrhea, indigestion, vomiting, sleepiness, and
trouble sleeping. In some patients, the results of
blood tests that measure how the liver or pancreas is
working may worsen.
SERIOUS SIDE EFFECTS AND WHAT TO DO
ABOUT THEM
Symptom / effect
Talk with your
doctor or
pharmacist
Stop
taking
drug and
call your
doctor or
pharmacist
Only if
severe
In all
cases
Lactic acidosis (high level of
lactic acid in the blood)
Symptoms:
Feeling very weak or tired
√
Unusual (not normal) muscle
pain
√
Trouble breathing
√
Stomach pain with nausea and
vomiting
√
Feeling cold especially in arms
and legs
√
Feeling dizzy or lightheaded
√
Fast or irregular heartbeat
√
Worsening hepatitis
(inflamed liver), liver
enlargement (hepatomegaly)
or fatty liver Symptoms:
Skin or the white part of eyes
turn yellow (jaundice)
√
Dark urine
√
Bowel movements (stools) turn
light in colour
√
Nausea
√
SERIOUS SIDE EFFECTS AND WHAT TO DO
ABOUT THEM
Symptom / effect
Talk with your
doctor or
pharmacist
Stop
taking
drug and
ll
Lower stomach pain
√
Loss of appetite for several
days or longer
√
This is not a complete list of side effects. For any
unexpected effects while taking BARACLUDE, contact your
doctor or pharmacist.
HOW TO STORE IT
Store BARACLUDE Tablets at room temperature, 15˚
to 30˚ C (59˚ F to 86˚ F). Keep the bottle in outer
carton to protect from light. They do not require
refrigeration.
Do not store BARACLUDE Tablets in a damp place
such as a bathroom medicine cabinet or near the
kitchen sink.
Keep the container tightly closed.
Discard BARACLUDE when it is outdated or no
longer needed by returning the unused portion to your
pharmacist for proper disposal.
Keep BARACLUDE and all medicines out of the
reach of children and pets.
General Information
Medicines are sometimes prescribed for conditions
other than those described in patient information
leaflets. Do not use BARACLUDE for a condition for
which it was not prescribed. Do not give
BARACLUDE to other people, even if they have the
same symptoms you have. It may harm them. The
leaflet summarizes the most important information
about BARACLUDE. If you would like more
information, talk with your healthcare provider. You
can also call Bristol Myers Squibb Canada at
telephone number 1-866-463-6267.

Page 43 of 43
REPORTING SUSPECTED SIDE EFFECTS
You can report any suspected adverse reactions
associated with the use of health products to the
Canada Vigilance Program by one of the following
3 ways:
• Report online at
www.healthcanada.gc.ca/medeffect
• Call toll-free at 1-866-234-2345
• Complete a Canada Vigilance Reporting
Form and:
- Fax toll-free to 1-866-678-6789, or
- Mail to: Canada Vigilance Program
Health Canada
Postal Locator 1908C
Ottawa, ON K1A 0K9
Postage paid labels, Canada Vigilance
Reporting Form and the adverse reactions
reporting guidelines are available on the
MedEffect
tm
Canada Web site at
www.healthcanada.gc.ca/medeffect.
Note: Should you require information related to the
manage
ment of side effects, contact your health
professional. The Canada Vigilance Program does
not provide medical advice.
MORE INFORMATION
This document plus the full product monograph,
prepared for health professionals, can be obtained by
contacting the sponsor, Bristol-Myers Squibb Canada
at 1-866-463-6267.
This leaflet was prepared by Bristol-Myers Squibb
Canada
Last revised: 23 September 2020
