
JOURNAL OF GEOPHYSICAL RESEARCH, VOL. 101, NO. D3, PAGES 6931-6951, MARCH 20, 1996
Chemical and optical properties of marine boundary layer
aerosol particles of the mid-Pacific in relation to sources
and meteorological transport
P. K. Quinn, V. N. Kapustin, • and T. S. Bates •
NOAA, Pacific Marine Environmental Laboratory, Seattle, Washington
D. S. Covert 1
Department of Atmospheric Sciences, University of Washington, Seattle
Abstract. Incorporating the direct effect of tropospheric aerosol on climate into global
climate models involves coupling the optical properties of the aerosol with its physical and
chemical properties. This coupling is strengthened if the optical, physical, and chemical
properties of the individual aerosol components are known as well as how these properties
depend on the air mass source and synoptic scale meteorology. To relate properties of the
aerosol components to air mass sources over a wide range of meteorological conditions, two
long latitudinal cruises were conducted in the central Pacific Ocean from 55øN to 70øS.
Submicron non-sea-salt (nss) SO4 = aerosol averaged about 35 to 40% of the submicron ionic
mass as analyzed by ion chromatography and 6% of the total ionic mass, while supermicron
nss SO4 = aerosol contributed about 1% to the total ionic mass. About 1% of the remaining
total ionic mass was composed of methanesulfonate and 90% was sea salt. Ionic mass
fractions of nss SO4 = aerosol were highest in regions having the longest marine boundary
layer residence times or the largest source of marine or continental gas phase precursors. The
calculated scattering by nss SOft aerosol was highest in these same regions due to the
dependence of scattering on particle size and the concentration of nss SO4 = in the submicron
size range. The calculated scattering by submicron sea salt was similar to that of the nss SO4 =
aerosol, indicating that its contribution to scattering in the marine boundary layer can be
significant or even dominant depending on its mass concentration. Mass scattering efficien-
cies for nss SO4 = at 30% RH ranged from 4.3 to 7.5 m 2 g-1 and for submicron sea salt from
2 -1
3.5 to 7.7 rn g-. Mass backscattering efficiencies for nss SO4 = ranged from 0.41 to
2 1 2 1
0.58 rn g- and for submicron sea salt from 0.33 to 0.63 rn g-. These values fall within the
same range as others reported previously for the marine atmosphere.
1. Introduction
The direct effect of tropospheric aerosol on climate results
from the scattering by particles of incoming shortwave radiation
with a portion of it being reflected back to space. Defining
climate forcing as an externally imposed change on the Earth's
heat balance implies that a knowledge of the radiative properties
of both natural and anthropogenic aerosol components are
needed to calculate any change in the reflected flux, AF R, due to
an anthropogenic perturbation in aerosol loading or optical
properties. Parameters needed to calculate AFR include the mass
concentration of each aerosol chemical component, the mass
scattering efficiency, or the light scattering efficiency per unit
mass of each aerosol component j, {•sp j, and the fraction of
scattered light that is directed upward, [3. '•he latter quantity can
be approximated through the measurement of the backscattered
,,
•Also at Joint Institute for the Study of the Atmosphere and
Ocean, University of Washington, Seattle.
Copyright 1996 by the American Geophysical Union.
Paper number 95JD03444.
0148-0227/96/95JD-03444505.00.
fraction, b, which is the fraction of scattered light that is
redirected to the backward hemisphere of the particle. Consider-
ation of b is useful as many values have been reported for a
variety of air mass types. The quantities b and [3 are equal for a
zenith Sun angle.
The dominant aerosol components present in the marine
boundary layer include nss SO4 = aerosol, sea-salt aerosol,
mineral dust, organic carbon species, and to a lesser extent,
elemental carbon. In general, a component is composed of
several chemical elements, compounds, or ions. Non-sea-salt
SO4 = aerosol found in the marine boundary layer can have
marine, volcanic, and anthropogenic sources. Therefore it can be
part of the natural aerosol system or it can be an anthropogenic
perturbation of that system such that it will contribute to climate
forcing by tropospheric aerosol. It may be derived from the
oxidation of biogenic dimethylsulfide (DMS) which is emitted
from the ocean surface [Andreae, 1986; Bates et al., 1987], the
oxidation of volcanic gas phase SO 2 [Stoiber et al., 1987], or the
long range transport of anthropogenic air masses from continen-
tal regions [Quinn et al., 1990]. The latter may involve transport
through the free troposphere [Clarke, 1993]. Impactor measure-
ments indicate that nss SO4 = aerosol mass is concentrated
primarily in the 0.1 to 1.0 pm diameter size range [e.g., Whitby,
6931

6932 QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
1978; Savoie and Prospero, 1982; Pszenny et al., 1989; Quinn
et al., 1993].
Sea-salt aerosol is derived from the evaporation of seaspray
droplets and hence is part of the natural marine aerosol system.
It dominates the mass of the supermicron particle size range in
the marine boundary layer but can also contribute a significant
amount of mass to the submicron size range [O'Dowd and
Smith, 1993]. The number concentration of sea-salt aerosol in
the submicron size range is small, however, often contributing
only 4 to 10% of the total number [Mclnnes et al., 1995a].
Particles containing mineral dust are produced by the
weathering of soils and rock as well as through industrial and
agricultural practices. These particles have diameters ranging
from less than 1 •m to 100 •m. Those with diameters up to
about 4 •m can be transported over long distances [Merrill et
al., 1994], so that at times they can contribute a significant
amount of mass to the marine boundary layer aerosol.
Organic carbon compounds in the marine boundary layer can
have natural marine and terrestrial sources as well as anthropo-
genic sources. The ocean is a source of particulate organic
species through the direct injection of biogenic surfactants from
bubble bursting processes [Monahan, 1986] or the emission of
gas phase precursors [Plass-Dulmer et al., 1995]. Gaseous
organic carbon species emitted by natural terrestrial sources and
anthropogenic combustion may be transformed to the aerosol
phase through oxidation or condensation and transported over
long distances to the marine atmosphere. To date, the organic
content of marine aerosol particles has not been well quantified
or speciated. The few data that have been reported indicate that
carbon-containing species can make up 4 to 15% of the total
marine aerosol mass with more than 90% of this mass in the
form of organic carbon [Rau and Khalil, 1993].
From the number-size distribution and an estimation of the
particle density and refractive index, the scattering and backscat-
tering coefficients, Osp and Ubsp, can be calculated for the total
aerosol with Mie theory. In addition, knowledge of the chemical
mass-size distribution of each aerosol component j allows for
the calculation of the scattering coefficient of that component,
or Usp, j. The scattering coefficient for the components then can
be used to determine the fractional contribution of the compo-
nent to scattering and backscattering by the aerosol as a whole
and to calculate the mass scattering efficiency of the component.
For aerosol component j the mass scattering efficiency is
defined as the component's scattering increment per mass
increment or
0 Osp,j ( 1 )
IgsP'/ - 0m. '
d
If the total mass of component j is not known, an individual
species can be used as an indicator for that component. For
example, the nss SO 4- ion can be used as an indicator to
represent the complete nss SO 4- aerosol component composed
of nss SO 4- and the mass of NH4* and H20 that is universally
associated with it under most atmospheric conditions.
The magnitude of the scattering and backscattering coeffi-
cients of each aerosol component depends on its size dependent
mass (or volume) concentration. The dependence of the scatter-
ing coefficient on particle size is shown in Figure 1 for a
wavelength of 0.55 Bm, particles of unit volume, and a refractive
index of 1.5-10-7i, which is near that of (NH4)2SO 4 and sea salt
at low relative humidity. For these conditions, on a mass or
volume basis, the scattering efficiency is lognormally distributed
with the most efficient size range for scattering occurring
between particle diameters of 0.2 and 1.0 Bm.
What this implies for scattering by atmospheric aerosols is
shown by comparing these calculated scattering efficiency size
distributions with size distributions of the atmospheric aerosol.
Figure 1 shows two measured size distributions, one from a
marine air mass (solid line with symbols) and one from a
continentally influenced air mass (dashed line with symbols).
For the marine case, the minimum between the submicron and
supermicron modes coincides with the maximum in the scatter-
ing distribution. Therefore scattering by the total aerosol will be
affected by both modes but largely controlled by the super-
micron mode due to its large mass concentration, in spite of the
decrease in the scattering coefficient with size. For the continen-
tal case, the submicron mode will have a larger effect on
scattering by the aerosol as a whole than it did in the marine
case, as its peak diameter is shifted toward the maximum in the
scattering distribution. The supermicron mode will have a
smaller effect, however, as it has a lower mass concentration.
Clearly, both the geometric volume mean diameter and the mass
concentration of an atmospheric aerosol component will
strongly affect the amount the component contributes to
scattering by the whole aerosol.
For a given aerosol mass concentration and number distribu-
tion, differences in chemical composition also will affect
scattering by the total aerosol. This effect is such that for
diameters between 0.2 and 0.3 gm, an increase in the real
portion of the refractive index from 1.4 (50% H2SO 4 / 50%
H20 ) to 1.5 ((NH4)2SO4) yields a 60% increase in O•p and a 50%
increase in Ubs p [Marshall 1994]. As a result, the degree of
neutralization of nss SO4 = aerosol by NH 3 (g) will affect
scattering by the total aerosol. Similarly, the addition of a
significant amount of sea salt, which also has a refractive index
near 1.5, to a H2SO 4 aerosol also will affect scattering by the
total aerosol. The backscattered fraction shows the largest
sensitivity to change in refractive index at a diameter near
1.0 pm [Marshall, 1994]. Hence b will change with wind speed
as the mass distribution of sea-salt aerosol changes above and
below 1.0 gm. The optical properties of the aerosol also will be
affected by the incorporation of a small amount of a strong
absorber such as elemental carbon. Such changes have yet to be
quantified, however.
As indicated in the above discussion, the size dependent
chemical composition and mass concentration of tropospheric
aerosol components determine the optical properties of the
aerosol. These size dependent aerosol parameters are, in turn, a
result of synoptic scale meteorology and air mass sources. Data
collected during two Radiatively Important Trace Species
(RITS) cruises in the central Pacific Ocean have allowed for an
analysis of the effect of meteorological transport on the number
distribution [Covert et al., 1995]. The number concentration in
the Aitken (particle diameter, Dp, of 0.01 to 0.1 gm) and
accumulation (Dp of 0.1 to 0.5 pm) modes was found to depend
on the relative importance of the transport of ultrafine
(<0.01 pm) and Aitken particles from the free troposphere to the
marine boundary layer versus the aging of Aitken and accumula-
tion mode particles within the boundary layer due to vapor
condensation and cloud processing. In middle- and high-latitude
regions, subsidence from the free troposphere was frequent and
aging processes were limited by the short amount of time the
aerosol spent in the boundary layer. As a result, the Aitken mode
dominated the number distribution but contributed little to total
mass or light scattering. In the tropics, more time in the bound-
ary layer and more frequent clouds led to size distributions with
roughly equal number in the Aitken and accumulation modes
and, accordingly, a greater mass in the accumulation mode.
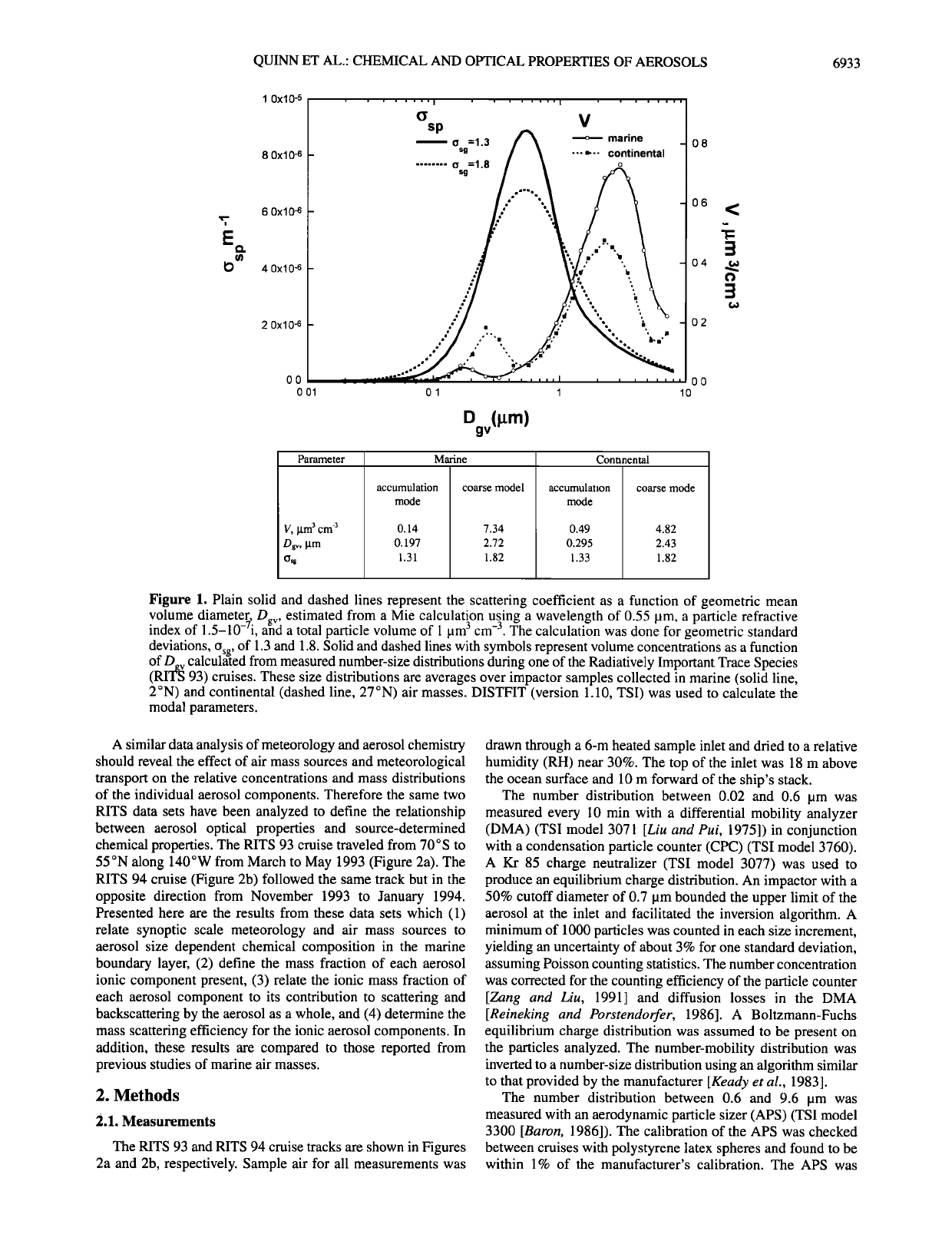
QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6933
1.0x10-5
8.0X10-6
6.0x10-6
4.0x10-6 -
2 0x10 -6 -
O0
001
i i ! . i , 1,1 ! [ ! ! , i ill I I . , ! . I.
Osp V
o =1.3 j• • marine - 0.8
_ sg / \ ...... continental
_ 0.6
.. -. • - 0.2
........... I , -'i•, O0
01 1 10
Dgv(,U,m)
Parameter Marine Continental
accumulation coarse model accumulation coarse mode
mode mode
V, gm 3 cm -3 0.14 7.34 0.49 4.82
Dg•, gm 0.197 2.72 0.295 2.43
o•g 1.31 1.82 1.33 1.82
Figure 1. Plain solid and dashed lines represent the scattering coefficient as a function of geometric mean
volume diameter D
index of 1.5-10-Yi, a• estimated from a Mie calculation using a wavelength of 0.55 pm, a particle refractive
a total particle volume of 1 pm 3 cm -3. The calculation was done for geometric standard
deviations, o , of 1.3 and 1.8. Solid and dashed lines with symbols represent volume concentrations as a function
of D._ v calcul•t•ed from measured number-size distributions during one of the Radiatively Important Trace Species
(RITES 93) cruises. These size distributions are averages over impactor samples collected in marine (solid line,
2øN) and continental (dashed line, 27øN) air masses. DISTFIT (version 1.10, TSI) was used to calculate the
modal parameters.
A similar data analysis of meteorology and aerosol chemistry
should reveal the effect of air mass sources and meteorological
transport on the relative concentrations and mass distributions
of the individual aerosol components. Therefore the same two
RITS data sets have been analyzed to define the relationship
between aerosol optical properties and source-determined
chemical properties. The RITS 93 cruise traveled from 70øS to
55 øN along 140øW from March to May 1993 (Figure 2a). The
RITS 94 cruise (Figure 2b) followed the same track but in the
opposite direction from November 1993 to January 1994.
Presented here are the results from these data sets which (1)
relate synoptic scale meteorology and air mass sources to
aerosol size dependent chemical composition in the marine
boundary layer, (2) define the mass fraction of each aerosol
ionic component present, (3) relate the ionic mass fraction of
each aerosol component to its contribution to scattering and
backscattering by the aerosol as a whole, and (4) determine the
mass scattering efficiency for the ionic aerosol components. In
addition, these results are compared to those reported from
previous studies of marine air masses.
2. Methods
2.1. Measurements
The RITS 93 and RITS 94 cruise tracks are shown in Figures
2a and 2b, respectively. Sample air for all measurements was
drawn through a 6-m heated sample inlet and dried to a relative
humidity (RH) near 30%. The top of the inlet was 18 m above
the ocean surface and 10 m forward of the ship's stack.
The number distribution between 0.02 and 0.6 pm was
measured every 10 min with a differential mobility analyzer
(DMA) (TSI model 3071 [Liu and Pui, 1975]) in conjunction
with a condensation particle counter (CPC) (TSI model 3760).
A Kr 85 charge neutralizer (TSI model 3077) was used to
produce an equilibrium charge distribution. An impactor with a
50% cutoff diameter of 0.7 pm bounded the upper limit of the
aerosol at the inlet and facilitated the inversion algorithm. A
minimum of 1000 particles was counted in each size increment,
yielding an uncertainty of about 3% for one standard deviation,
assuming Poisson counting statistics. The number concentration
was corrected for the counting efficiency of the particle counter
[Zang and Liu, 1991] and diffusion losses in the DMA
[Reineking and Porstendorfer, 1986]. A Boltzmann-Fuchs
equilibrium charge distribution was assumed to be present on
the particles analyzed. The number-mobility distribution was
inverted to a number-size distribution using an algorithm similar
to that provided by the manufacturer [Keady et al., 1983].
The number distribution between 0.6 and 9.6 }am was
measured with an aerodynamic particle sizer (APS) (TSI model
3300 [Baron, 1986]). The calibration of the APS was checked
between cruises with polystyrene latex spheres and found to be
within 1% of the manufacturer's calibration. The APS was

6934 QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
(a)
i'
..... RITS 1993
'-e.
160 ø 180 ø 160 ø 140 ø 120 ø 1 O0 ø 80 ø 60 ø
60øN
20øN
o
20øS
40 ø
- 60oS
rnb
73 ø
40ow 960 1000 1040
Figure 2. Composite surface synoptic maps of the Pacific for (a) RiTS 93 and (b) RITS 94. Several surface maps
were combined to indicate the changing meteorological conditions encountered by the ship along the cruise track.
The cruise tracks and location of impactor samples also are indicated. Measured sea level pressures along the
cruise track are shown in the sidebar.
operated at sample and sheath airflow rates used by the manu-
facturer for the calibration. All flow rates were checked in the
field and adjusted to better than 1% on a daily basis. The sheath
air for both sizing systems was cleaned with absolute particle
and charcoal filters. The APS diameters were converted to
geometric diameter by dividing by the square root of a particle
density of 1.9 gcm -3, corresponding to that of sea salt. All DMA
and APS results are plotted as the incremental distribution of
number concentration, AN/AlogDp, versus log of geometric
particle diameter at 30% RH.
A seven-stage multijet cascade impactor [Berner et al., 1979]
was used to collect samples for the determination of mass
distributions of CI-, Br-, NO3-, SOn =, methanesulfonate or
+2
MSA-, Na +, NH4 +, K + , Mg +2, and Ca . Samples were collected
only when the concentration of particles greater than 15 nm in
diameter indicated the sample air was free of contamination
from the ship. In addition, samples were taken only when the
wind speed was greater than 3 m s -• and the wind direction was
forward of the beam. The stages of the impactor had 50%
aerodynamic cutoff diameters, Ds0, of 0.125, 0.25, 0.5, 1.0, 2.0,
4.0, and 8.0 lam. For comparison to measurements of the number
distribution the impactor aerodynamic diameters were converted
to geometric diameters by dividing by the square root of an
assumed density of 1.7 gcm -3, which approximates that of
ammoniated sulfate salts and sea salt at a relative humidity of
30%. The resulting 50% geometric cutoff diameters are 0.096,
0.19, 0.38, 0.77, 1.5, 3.1, and 6.1 gm. All impactor results are
plotted as Am/AlogDp versus log of geometric particle diameter
at 30% RH.
Tedlar films were used as the collection substrate for the six
largest stages and a Millipore Fluoropore filter (1.0-gm pore
size) was used for the smallest stage. The Millipore filter has a
collection efficiency of 99% or greater for particles with
diameters larger than 0.035 gm [Liu and Lee, 1976]. An
impaction stage (Ds0 • 10 gm) at the inlet of the impactor was
covered with silicone grease to prevent bouncing of large
particles onto the downstream stages.
To avoid sample artifacts due to contaminated substrates,
films were cleaned in an ultrasonic bath in 10% H202 for 30
min, rinsed 6 times in distilled, deionized water, and dried in an
NH 3- and SO2-free glove box. All handling of the substrates was
done in the glove box. Blank levels were determined by loading
the impactor with the substrates and deploying it at the sampling
site for the length of a typical sampling period without pulling
air through it. Following collection, the blanks were treated in
an identical manner as the sample substrates. On average, the
Na +, NH4 +, K +, Mg +2, Ca +2, NO3-, and SO4 = blanks were 10, 30,
40, 8, 70, 5, and 8% of the sample values, respectively. The CI-,
Br-, and MSA-blanks were below detection limit.
The time period of impactor sampling ranged from 12 to 24
hours. After sample collection the material on the films and filter
was extracted by first wetting with 1 mL of methanol and then

QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6935
. 13• 14 • :
,%..::'... ø• . . . 161F' .... .... ::..
I I I I I I 1
160 ø 180 ø 160 ø 140 ø 120 ø 1 O0 ø 80':' 60':'
• 60ON
- 40 ø
- 20ON
- 0 o
20øS
40 ø
60øS
73 ø
40øW
960
rnb
1000 1040
Figure 2. (continued)
adding 5 mL of distilled deionized water and sonicating for 15
min. Extracts were analyzed by ion chromatography. The cation
analysis was done with a Dionex CS-12 column, 20-mM MSA
eluant, and deionized water regenerant using the self-regenerat-
ing Dionex CSRS-1 suppressor system. Anion analysis was
done with a Dionex AS-4A column, 0.76-mM NaHCO3/2.0-mM
Na2CO 3 eluant, and 12.6-m]V/H2SO 4 regenerant. MSA-analysis
was performed with a Dionex AS-4 column, a 5-mM NaOH
mobile phase to elute the weak organic acids followed by 100-
mM NaOH to elute the stronger acids, and 12.6-mM H2SO 4
regenerant. Non-sea-salt SO4 = concentrations were calculated
from Na + concentrations and the molar ratio of sulfate to sodium
in seawater of 0.0603 [Holland, 1978].
Measurements of the scattering coefficient, Osp,meas, were
made at a wavelength of 0.55 gm with an integrating nephelom-
eter [Bodhaine et al., 1991] over a scattering angle, 0, where
8 ø _< 0 _< 168 ø. Every 3 to 4 days the nephelometer was cali-
brated with the Rayleigh scattering value of CO 2 of 2.61 times
air and was zeroed with particle-free air.
Ancillary measurements included surface temperature, dew
point, wind speed, wind direction, and rawinsonde data.
Rawinsonde balloons were launched at standard times of 0000
and 1200 GMT. Air mass back trajectories were calculated for
up to 12 days using the hybrid single-particle Lagrangian
integrated trajectories model, HY-SPLIT, based on wind fields
generated by the medium-range forecast (MRF) model [Draxler,
1992]. Trajectories were terminated at the ship's location at
1000 mbar. Surface and upper air maps were obtained from the
National Weather Service archives to aid in the analysis of
meteorological conditions and air motion.
2.2. Model Calculations
2.2.1. Calculation of scattering and backscattering by the
total aerosol. The first step in calculating the light scattering
(backscattering) due to each aerosol component was to test the
ability of the Mie model to estimate accurately the scattering
characteristics of the aerosol as a whole over all particle sizes
and chemical components. To calculate the scattering due to the
total aerosol, the Mie model was applied to the measured
number distributions and the density and refractive index were
derived from the measured chemical composition. These
calculated scattering values then were compared to those
measured directly with the nephelometer. The results of the
comparison are discussed in section 3.4. Backscattering values
also were calculated to derive the backscattered fraction b.
The model calculations described briefly here are presented
in greater detail in Marshall [ 1994]. The scattering and back-
scattering efficiencies, Qsp and Qbsp, were obtained at discrete
particle sizes by integrating over the scattered intensity function
from 8 ø to 168 ø and 90 ø to 168 ø, respectively [Bohren and
Huffman, 1983]. These efficiencies were then summed over the
measured number distribution to yield the calculated scattering,
Osp,calc, and backscattering, Obsp,calc , coefficients using

6936 QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
Osp;bsp: E Qsp;bsp (Oi,g',n) •Z •
aN
AlogD i
4 AlogD i
(2)
where D i is the geometric diameter of the measured particle size
increment. Particles are assumed to be spherical, 3, is the
wavelength of incident light, and n is the particle refractive
index. Even though the particles are subjected to decreasing RH
as they pass through the heated sampling inlet, they are expected
to remain spherical solution droplets due to hysteresis in the
crystallization of the hygroscopic nss SO4 = and sea-salt aerosol
components. Therefore the uncertainty associated with the
assumption of spherical particles is expected to be small.
The number distributions used in (2) were obtained by
averaging the measured DMA and APS data over the impactor
sampling times. Particles were assumed to be homogeneous
spheres distributed as an external mixture of two components:
a nss SO4 = aerosol composed of nss SO4 =, NH4 +, and H20, and
a sea-salt aerosol composed of Na +, K +, Mg +2, Ca +2, CI-, Br-,
NO3-, and sea-salt SOft. The assumption of an external mixture
is supported by the clear size separation of nss SO4 = and sea-salt
aerosol into the submicron and supermicron size ranges (see
section 3.2) and by individual particle analysis with an analytical
electron microscope [Mclnnes et al., 1995a].
The particle number in any size increment was divided
between the components based on the relative mass of the
components determined from chemical analysis of the impactor
stages. The water mass associated with the nss SO4 = aerosol
component was assumed to be equivalent to that associated with
an ammoniated sulfate salt at 30% RH neutralized to the degree
indicated by the measured NH4 + to nss SO4 = molar ratio. This
RH corresponds to the operating conditions of the nephelometer,
sizing instrumentation, and impactor. Water was assumed to be
a small fraction of the sea-salt component mass and was not
included in the calculation. In cases where one or more of the
sea-salt ion concentrations were unavailable, a concentration
was calculated using the molar ratio of the ion to Na + in
seawater [Holland, 1978].
The sea-salt mass was assigned a constant refractive index of
1.5-10-8i [Kent et al., 1983] and a density of 1.9 g cm -3. The
density and refractive index of the nss sulfate component were
determined by partitioning it into either a mixture of H2SO 4 and
NH4HSO 4 or a mixture of NH4HSO 4 and (NH4)2SO 4, according
to the measured NH4 + to nss SO4 = molar ratio for each sample
and impactor stage within that sample. The density of each
mixture was derived from solution data for H2SO 4 [Bray, 1970],
NH4HSO 4 [Tang and Munkelwitz, 1994], and (NH4)2SO 4 [Tang
and Munkelwitz, 1991 ]; values ranged from 1.41 to 1.77 g crn -3.
The imaginary refractive index of the sulfate component was
given a constant value of 10-7i [Kent et al., 1983]. The real
portion of the refractive index was estimated using the molar
refraction method of Stelson [ 1990] and values of the electrolyte
molar refraction from Tang and Munkelwitz [1994]; values
ranged from 1.40 to 1.52.
The Mie calculations were performed at a wavelength of
0.55 }am corresponding to the central value of the nephelometer
wavelength response. Sensitivity calculations using a gaussian
wavelength response centered at 0.55 pm yielded scattering and
backscattering coefficients within a few percent of the single
wavelength calculations [Marshall, 1994].
There are uncertainties associated with the measurements
which contributed to the uncertainty of Osp,cal c and Obsp,calc. The
accuracy of the calculated scattering coefficients depends on the
completeness of the characterization of the chemical composi-
tion of the aerosol which in this case was determined by ion
chromatography. To determine what portion of the aerosol mass
was accounted for by the ion chromatography analysis, filters
which collected the submicron fraction of the aerosol were
analyzed gravimetrically and the resulting mass was compared
to the mass from the ion chromatography (IC) analysis. Several
precautions were taken to maintain the accuracy and precision
of the gravimetric measurements. The balance was checked
every tenth sample for instrumental drift with calibration
weights. In addition, it was housed in a humidity- and
temperature-controlled glove box located in a class 100 clean
room. The resulting uncertainty was +20%. Further details about
the gravimetric analysis can be found in the work of Mclnnes et
al. [ 1995b].
The average and standard deviation of the IC mass relative to
the gravimetrically determined mass over all RITS 93 and RITS
94 samples was 80 + 20%. It was difficult to correct for the
missing mass in the scattering calculations, since it was not
known which particle diameters within the submicron size range
it was associated with. Therefore it was assumed that all of the
scattering was due to the nss SO4 = and sea-salt components. In
cases where the majority of the mass was nss SO4 = and sea salt,
this assumption does not add any uncertainty to the scattering
Calculations. In cases where the IC mass is lower than the
gravimetrically analyzed mass, the scattering due to nss SO4 =
and sea salt will be overpredicted in proportion to the amount of
missing mass. The largest error associated with this assumption
would be a 20% overprediction of scattering by nss SO4 = or sea
salt in a certain size range.
Other uncertainties include statistical fluctuation in the
average number concentration and instrumental errors in particle
sizing and counting due to drift of the airflow in the DMA and
APS. An error propagation analysis was carried out to estimate
the combined uncertainty of the calculated scattering. This
analysis indicated an estimated uncertainty of about +20% and
-13% in the calculation of both Osp,cal c and Obsp,calc [Marshall,
1994].
2.2.2. Calculation of scattering and backscattering by the
aerosol components. Calculations were performed to determine
the fraction of the total scattering and backscattering due to the
nss SOn = and sea-salt aerosol components. The number concen-
tration at each point in the measured number distribution was
divided between the two components according to the measured
ionic mass composition for the corresponding impactor interval.
In this way the more detailed number distribution information
was preserved. For example, the nss SOn = aerosol number
distribution was estimated from
A l•gDp SO4'aer
= / mso4'aer / •)SO4'aer
mso4,aer/0SO4,aer + mseasalt/0seasalt
AN
AlogDp
(3)
where mso4,aer and mseasal t are the nss SO4 = and sea-salt aerosol
masses, pSO4,aer and Pseasalt are the nss SO4 = and sea-salt aerosol
densities, and _AN/AlogDp is the measured number distribution.
The nss SO 4- aerosol light scattering and backscattering
coefficients were calculated by substituting values of
(A N/ AlogDp)sO4, aer into (2). The sea-salt aerosol number

QUINN ET AL.' CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6937
distribution and scattering (backscattering) coefficients were
calculated similarly.
Equation (3) assumes that the chemically analyzed species
account for the total measured number concentration and that
the aerosol is an external mixture. A comparison of IC and
gravimetrically analyzed mass indicate that the IC-identified
aerosol components make up about 80% of the submicron
aerosol mass [Mclnnes et al., 1995b]. Therefore in instances
where other components are present, equation (3) will overesti-
mate by up to 20% the number distribution as well as the
scattering of the ionic aerosol components. No attempt was
made to correct for this as it is not known how the unaccounted
for mass is distributed with size. As stated above, the clear size
separation of nss SO4 = and sea-salt aerosol into the submicron
and supermicron size ranges (see section 3.2.) and results of
individual particle analysis with an analytical electron micro-
scope [Mclnnes et al., 1995a] support the assumption of an
externally mixed aerosol.
3. Results
3.1. Meteorology and Calculated Air Mass
Back Trajectories
A composite of surface synoptic maps representing condi-
tions during RITS 93 and RITS 94 are shown in Figures 2a and
2b, respectively, along with the cruise track and locations where
impactor samples were collected. These figures are composites
in the sense that they combine many surface weather maps to
best approximate the synoptic features and motions encountered
by the ship at each latitudinal region along the cruise track.
Selected trajectories that are representative of both cruises are
shown in Figure 3. Major synoptic features were similar for the
two cruises.
Mean MBL
Residence Time
High-L atitude
Synoptic Systems
< 3 Days
Mid-Latitude
High pressure
3-5 Days
Tropical Depression
>5 Days
Mid-Latitude
high pressure
region
High-Latitude
synoptic system
<3 Days
: 8
• 6
..... IT. CZ ..........
: 5
.
4
ß .-0 RITS 1993
--i-i RITS 1994
(• 7 Apr 93
• 21 Dec 93
(•) 11 Apr93
• 22 Apr 93
• 5 Dec 93
(•) 25 Apr 93
(• 29 Apr 93
• 29 Nov 93
60øN
45 ø
30 ø
15 ø
o
15øS
30 ø
60 ø
150 ø 120 ø 90 ø 60øW
Figure 3. Back trajectories representative of those calculated for the RITS 93 and RITS 94 cruises. The
trajectories end at the ship's position; each point represents 1 day back in time. The portion of each trajectory
at less than 900-mbar pressure is plotted as a thick line, while the portion above that level is plotted as a dashed
line. The 900-mbar surface was taken as the average height of the marine boundary layer based on analysis of
the shipboard rawinsonde data. The sidebar indicates the marine boundary layer residence time which is defined
as the length of time the air parcel and the aerosol it contained spent below 900 mbar prior to being sampled on
the ship.

6938 QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
At high latitudes (>45 o or 50ø), in both the northem and the
southern hemispheres, the passage of low-pressure systems from
west to east occurred every few days. Sea level pressures in
these regions were the lowest measured along both the RITS 93
and the RITS 94 cruise tracks. Subsidence of air from above the
marine boundary layer height of about 1000 m was associated
with frontal passages as indicated by trajectories (Figure 3,
trajectory 1) and increases in the ultrafine and Aitken particle
concentrations [Covert et al., 1995]. As a result, these regions
had short marine boundary layer residence times of about 1 day.
Here, the marine boundary layer residence time of an air parcel
and the aerosol it contains is defined as the length of time the
parcel spent below 900 mbar prior to being sampled on the ship.
This residence time was estimated from the calculated back
trajectories.
Belts of strong high-pressure systems moving from west to
east existed in the midlatitudes from about 40øS to 20øS and
from 20øN to 40øN. The latitudes given are approximate as the
position of these systems varied with season and year. The
location of the ship relative to the high-pressure systems
determined the trajectories of the air being sampled. During
RITS 93, as the ship traveled north from 50øS to 20øS, it
remained within one well-developed high. The transit of the ship
from the low-pressure region of the higher latitudes to this high-
pressure region in the midlatitudes is evidenced by the sharp
increase in sea level pressure from 960 to 1030 mbar (Figure
2a). On the equatorward side of the high, calculated trajectories
were from the southeast (Figure 3, trajectory 3). This air was
transported from the free troposphere at 55øS through the
southeast sector of the high. Subsidence followed by transport
along the edge of the high resulted in boundary layer residence
times of about 4 days. On the poleward side of the high, air
subsided from the free troposphere and was transported along
the southern edge of the high. Here, boundary layer residence
times were about 3 days.
During RITS 94, while traveling from north to south, the ship
entered the southern hemisphere midlatitudes as a low-pressure
system near Tahiti formed between two highs. The ship moved
with the low-pressure system from about 20øS to 40øS. Hence
there is only a gradual decrease in the measured sea level
pressure as the ship moved from 45øS to 60øS. Trajectories
(Figure 3, trajectory 2) indicate that the sampled air was
transported from the northeast along the surface to this low-
pressure region. The sampled air had a boundary layer residence
time of up to 5 days.
In the northern hemisphere midlatitudes during RITS 93 the
ship traveled through a pseudostationary high from about 20øN
to 40øN. On the equatorward side of the high the sampled air
was transported along the edge of the high from the northeast
with a boundary layer residence time of 3 to 5 days (Figure 3,
trajectory 7). High number and sulfate mass concentrations
indicate that this air had been continentally influenced prior to
its arrival at the ship. During RITS 94, as the ship moved from
north to south, a low-pressure system was encountered at 55øN
and again near 45øN. The ship traveled with the second low
until about 20 øN. Trajectories (Figure 3, trajectory 8) were of a
more marine origin than for RITS 93, with sampled air having
been transported from the northwest with a boundary layer
residence time of at least 2 days.
The well-developed midlatitude highs in both the northern
and the southern hemispheres led to a tropical depression
between about 20øS and 20øN. This low-pressure belt is
indicated by the decrease in measured sea level pressure for both
RITS 93 and RITS 94. Within this belt, air flowing around the
high-pressure systems resulted in relatively stable and consistent
marine boundary layer trade wind flow for both cruises. Easterly
trajectories (Figure 3, trajectories 4, 5, 6) indicated that the
sampled air masses had boundary layer residence times of up to
or more than 7 days. Based on atmospheric CO concentrations,
the Intertropical Convergence Zone (ITCZ) was located at about
2 øN during RITS 93 and between 5 o and 10øN during RITS 94
[Bates et al., 1995].
3.2. Size Distributions
Number- and chemical mass-size distributions were analyzed
for the regions described above as they each give information
about the aerosol. Modal features of the number distribution
reveal information about the age of the aerosol or degree to
which the aerosol has been affected by coagulation, condensa-
tion, and cloud processes [Covert et al., 1995]. Chemical mass
distributions yield information about the aerosol source. Plots of
average number distributions for different latitudinal regions and
the variability observed over these regions are shown in Figures
4 (RITS 93) and 5 (RITS 94). The measured number distribution
data extend to a diameter of 10 pm. They are only shown to 0.6
pm, however, as larger diameters are indistinguishable from the
x axis on the scale used for the y axis. Examples of data for the
entire size range are shown as volume-size distributions in
Figure 6. Particle number, nss SO4 =, and Na + size distributions
are presented as contour plots of concentration versus size and
sample number (or latitude) in Figures 7 through 9. These
contour plots allow for a more detailed presentation of the data
as a time series than would be possible with a sequence of two-
dimensional plots.
The Aitken mode dominated the number distribution in the
high latitudes (>45øS and >45 øN) during both RITS 93 (Figures
4a, 4f, and 7a) and RITS 94 (Figures 5a, 5f, and 7b). Figures 4a,
4f, 5a, and 5f also indicate the high variability in the Aitken
mode number concentration in these regions. This type of
distribution is indicative of intrusion of air from the upper
troposphere into the boundary layer [Clarke, 1993; Covert et al.,
1995]. Aerosol growth from the Aitken to the accumulation
mode size range due to vapor condensation and cloud processing
is thought to occur mainly in the boundary layer and in stratus
and stratocumulus clouds [Hegg et al., 1992; Quinn et al., 1993;
Hoppel et al., 1994]. The short boundary layer residence times
associated with these distributions prevented the increase of
number and nss SO4 = mass in the accumulation mode (Figures
8a and 8b).
The difference in the position of the ship relative to the
midlatitude highs in the southern hemisphere for RITS 93 and
RITS 94 is reflected in a difference in the measured number
distributions (Figures 4b and 5b). During RITS 93 the ship
moved through one high-pressure system between 42øS and
20øS. The free tropospheric source of the sampled air is
indicated by the Aitken modal diameter, which is less than 20
nm [Covert et al., 1995], and dominance of the Aitken mode
(Figures 4b and 7a). Further north, from 20øS to 18øS, the
sampled air had spent slightly more time in the boundary layer,
so that the Aitken modal diameter was larger than 20 nm (Figure
4c). The relatively short boundary layer residence time led to
low accumulation mode nss SO4 = mass concentrations (Figure
8a).
During RITS 94 the ship was moving with a low-pressure
system within this same region. From both 42øS to 20øS (Figure
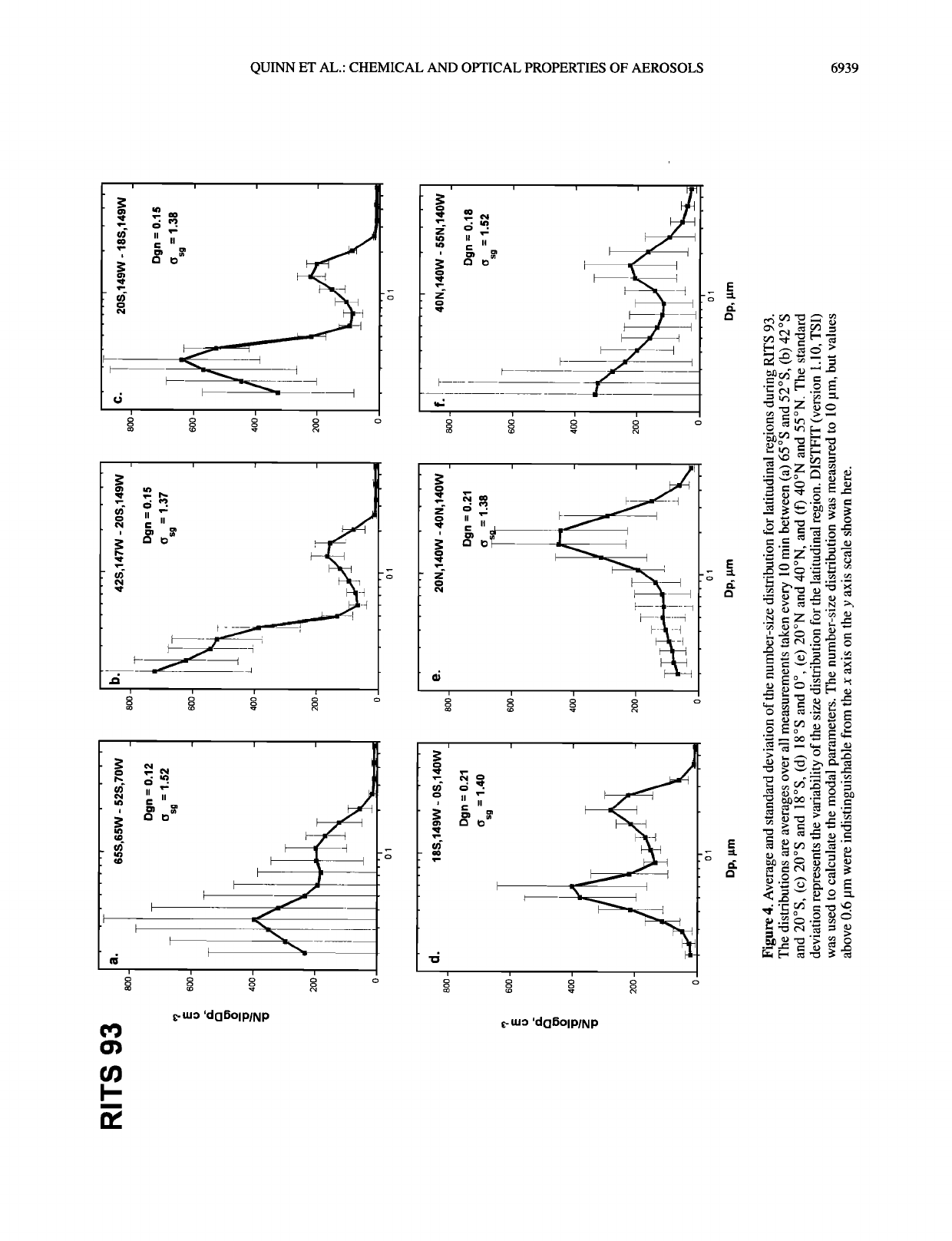
QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6939
II
II
I
C: II
I I I I I I I I
0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0
I I I I I I I l
0 0 0 0 0 0 0 0 0
0 0 0 0 0 0 0 0
I I I I I I
II cn
•,• o
I
,
! I I I I I
0 0 0 0 0 0 0
0 0 0 0 0 0
I 1
0 0
0 0
=.wo 'dal•olPlNP
•:-wo 'dal•olPlNP
E
E
E

6940 QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
II II
i i i i
,/
i i i i
o o o o
-q
I
i-
I
o
E
II II •
I I I i
o o o o
o o o o
II
II
1 i i i
o o o o
o o o o
E
s- tuo 'da15olPlNP =- tuo 'da15olPlNP
QUINN ET AL.' CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6941
10
0.1
0.01
1E-3
1E-4
---o-- - R94 46S - 18S
-R94 15S- 5N
- R94 5N - 40N
........ R93 45S-19S
....... -R93 19S-10N
-R93 10N-40N
! i i i i i i , I ! ! ! , , ! , i I . i , , . , i . I
0.01 0 1 1 10
D, !.tin
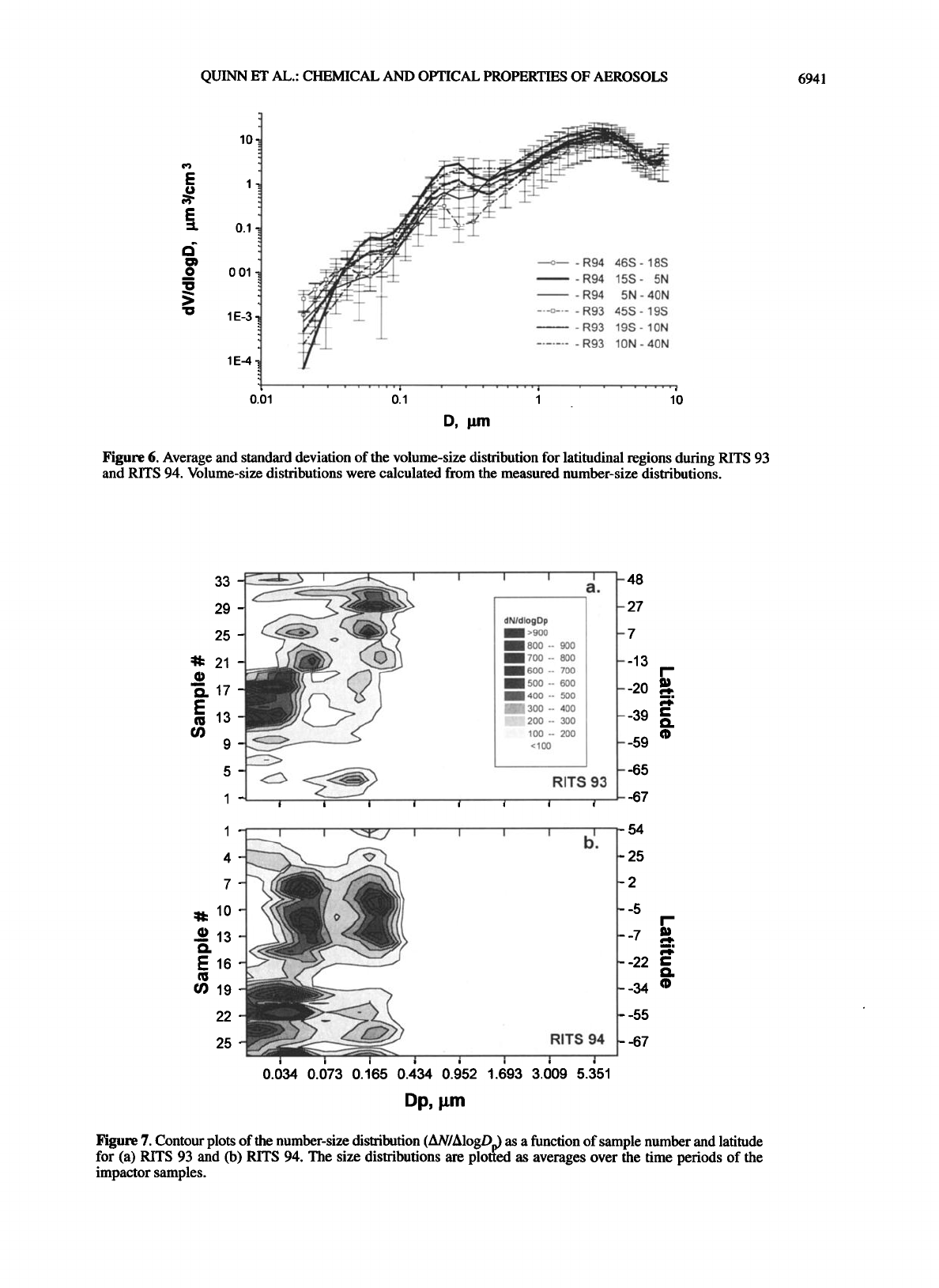
Figure 6. Average and standard deviation of the volume-size distribution for latitudinal regions during RITS 93
and RITS 94. Volume-size distributions were calculated from the measured number-size distributions.
29
25
• 21
•.17
• 13 .............. ..,.._,,...?•..i. 11 : .. :'. '
. - ---:---i-- .... ::::::::::::::::::::::::
........
1 I I I I
I I I I
a,
i i
dNIdlogDp
• >900
1800 -- 900
1700 -- 800
1600 -- 700
1 500 -- 600
.__11•_ _ 400 -- 500
-"'"'":•'½•-.';•*..-4• 300 -- 400
100 -- 200
<100
RITS 93
i i
1 I I I I b !.
4
7
:1:1= 10
• 13
L'• i• .:?i i'•-i: !i•:.:..:. 5:.., :. "•i•!!::::•::.':'.'.•. :.:-: :: .• :"
• 19 :':-':':'""i.11 ..... "i'•:'•'.:•-.-:.?'::u.•: .....
22
25 RIIS 94
! I I I I I I I
0.034 0.073 0.165 0.434 0.952 1.693 3.009 5.351
Dp, gm
- 48
- 27
-7
--39 1=
I3.
- -59
- -65
- -67
- 54
-25
-2
--5
--22 1=
I3.
--34 •
- -55
- -67
Figure 7. Contour plots of the number-size distribution (AN/AlogDp) as a function of sample number and latitude
for (a) RITS 93 and (b) RITS 94. The size distributions are plotted as averages over the time periods of the
impactor samples.
6942 QUINN ET AL.' CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
a. RITS 93, SO4
33 -1.--4---•'""'"••'- ::'•••• ...................................... ""• "••••••"'.-•:'i...!• • • •
•!E?.--.:::-•.:...: :.,.,.,•: .......... •..:
, •:•.• •t.• • -•'.-
29 '• ........ ::
25 .... '?" '•'• .... ' ............ '
..•:::.. •:• ............ ;'.•. ,:.•:=•:" •
• •7
• ::. :?:. .••,.•.•:•.•...•.............:.:::.:::::::•,;::.:;•:•.-••
.............
m •3
9 •-.:.:...-.-......-. - :.-- .•
5 •/ •
! I
I I I
_
7 - ) .......... •' '" •"•• ........ •'•"'•"'"'•""•::•,,:,.:..
:g::: •,•. ß -r:•,,•:'•; ......
)..i• I
19 .............. •.....•.•.••.•..'...
25
I I I I
0.077 0.135 0.271 0.542
:#: 10-
•. 13-
E
• 16-
I I
I
1.085
I I
RITS 94, SO4
pglm$1dlogDp
• >1.0
._• 0.8 -- 1.0
.:•o.6 -- 0.8
•-71•:::;.!::•: 0.2 -- 0.4
< 0.2
I
2.17
• - 48
-27
-7
- -13
--20 _•,
--39
- -59
- -65
- -67
- 54
-25
-2
---5
--7
--22
--34
- -55
- -67
4.34
Dp, i•m
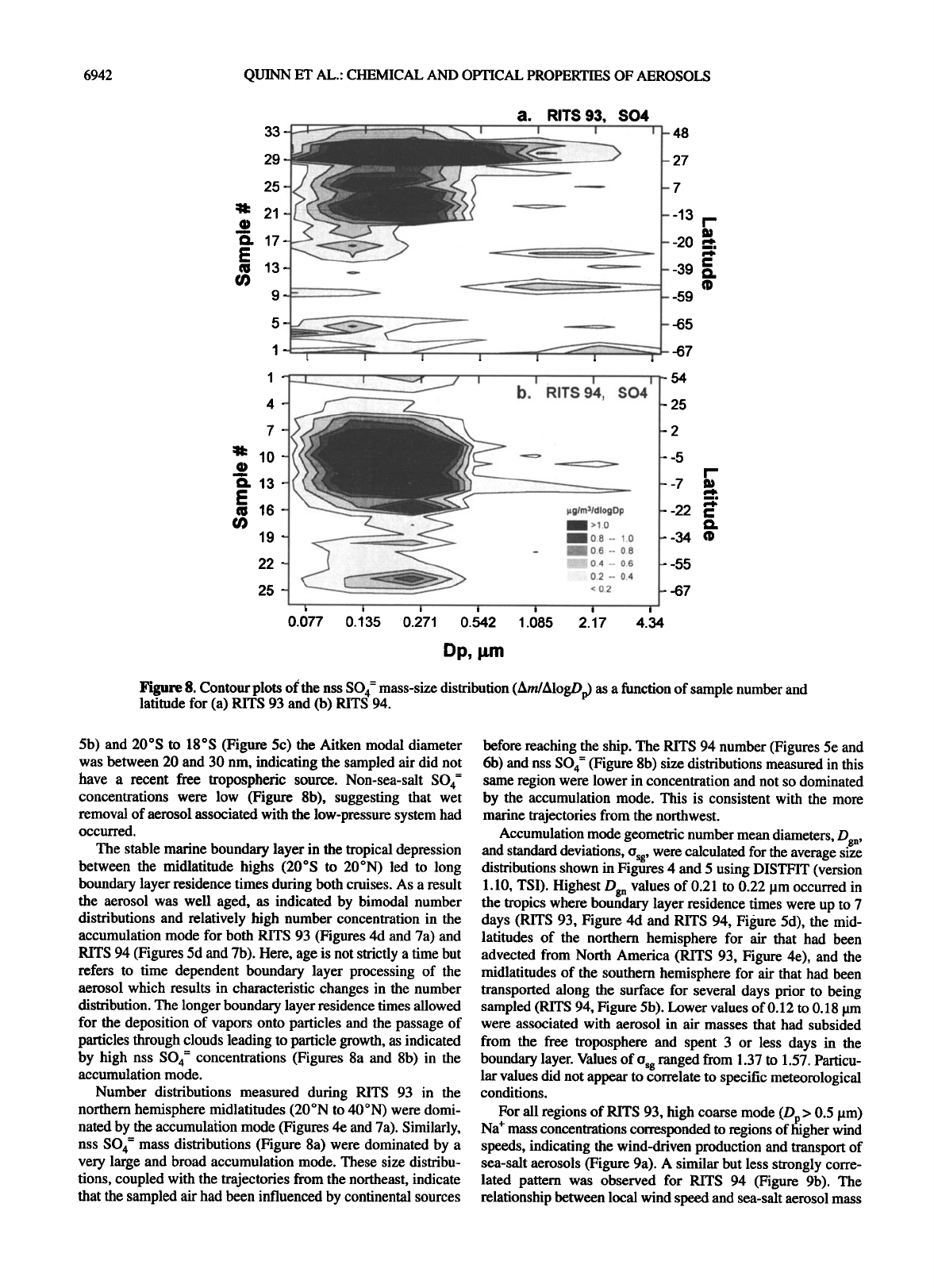
Figure 8. Contour plots of the nss SO4 = mass-size distribution (Am/AlogDp) as a function of sample number and
latitude for (a) RITS 93 and (b) RITS 94.
5b) and 20øS to 18øS (Figure 5c) the Aitken modal diameter
was between 20 and 30 nm, indicating the sampled air did not
have a recent free tropospheric source. Non-sea-salt SO 4-
concentrations were low (Figure 8b), suggesting that wet
removal of aerosol associated with the low-pressure system had
occurred.
The stable marine boundary layer in the tropical depression
between the midlatitude highs (20øS to 20øN) led to long
boundary layer residence times during both cruises. As a result
the aerosol was well aged, as indicated by bimodal number
distributions and relatively high number concentration in the
accumulation mode for both RITS 93 (Figures 4d and 7a) and
RITS 94 (Figures 5d and 7b). Here, age is not strictly a time but
refers to time dependent boundary layer processing of the
aerosol which results in characteristic changes in the number
distribution. The longer boundary layer residence times allowed
for the deposition of vapors onto particles and the passage of
particles through clouds leading to particle growth, as indicated
by high nss SO 4- concentrations (Figures 8a and 8b) in the
accumulation mode.
Number distributions measured during RITS 93 in the
northern hemisphere midlatitudes (20øN to 40øN) were domi-
nated by the accumulation mode (Figures 4e and 7a). Similarly,
nss SO 4- mass distributions (Figure 8a) were dominated by a
very large and broad accumulation mode. These size distribu-
tions, coupled with the trajectories from the northeast, indicate
that the sampled air had been influenced by continental sources
before reaching the ship. The RITS 94 number (Figures 5e and
6b) and nss SO 4- (Figure 8b) size distributions measured in this
same region were lower in concentration and not so dominated
by the accumulation mode. This is consistent with the more
marine trajectories from the northwest.
Accumulation mode geometric number mean diameters, D•n,
and standard deviations, o•, were calculated for the average size
distributions shown in Figures 4 and 5 using DISTFIT (version
1.10, TSI). Highest D•n values of 0.21 to 0.22 •m occurred in
the tropics where boundary layer residence times were up to 7
days (RITS 93, Figure 4d and RITS 94, Figure 5d), the mid-
latitudes of the northern hemisphere for air that had been
advected from North America (RITS 93, Figure 4e), and the
midlatitudes of the southern hemisphere for air that had been
transported along the surface for several days prior to being
sampled (RITS 94, Figure 5b). Lower values of 0.12 to 0.18 •m
were associated with aerosol in air masses that had subsided
from the free troposphere and spent 3 or less days in the
boundary layer. Values of o• ranged from 1.37 to 1.57. Particu-
lar values did not appear to correlate to specific meteorological
conditions.
For all regions of RITS 93, high coarse mode (Dp > 0.5 •m)
Na* mass concentrations corresponded to regions of higher wind
speeds, indicating the wind-driven production and transport of
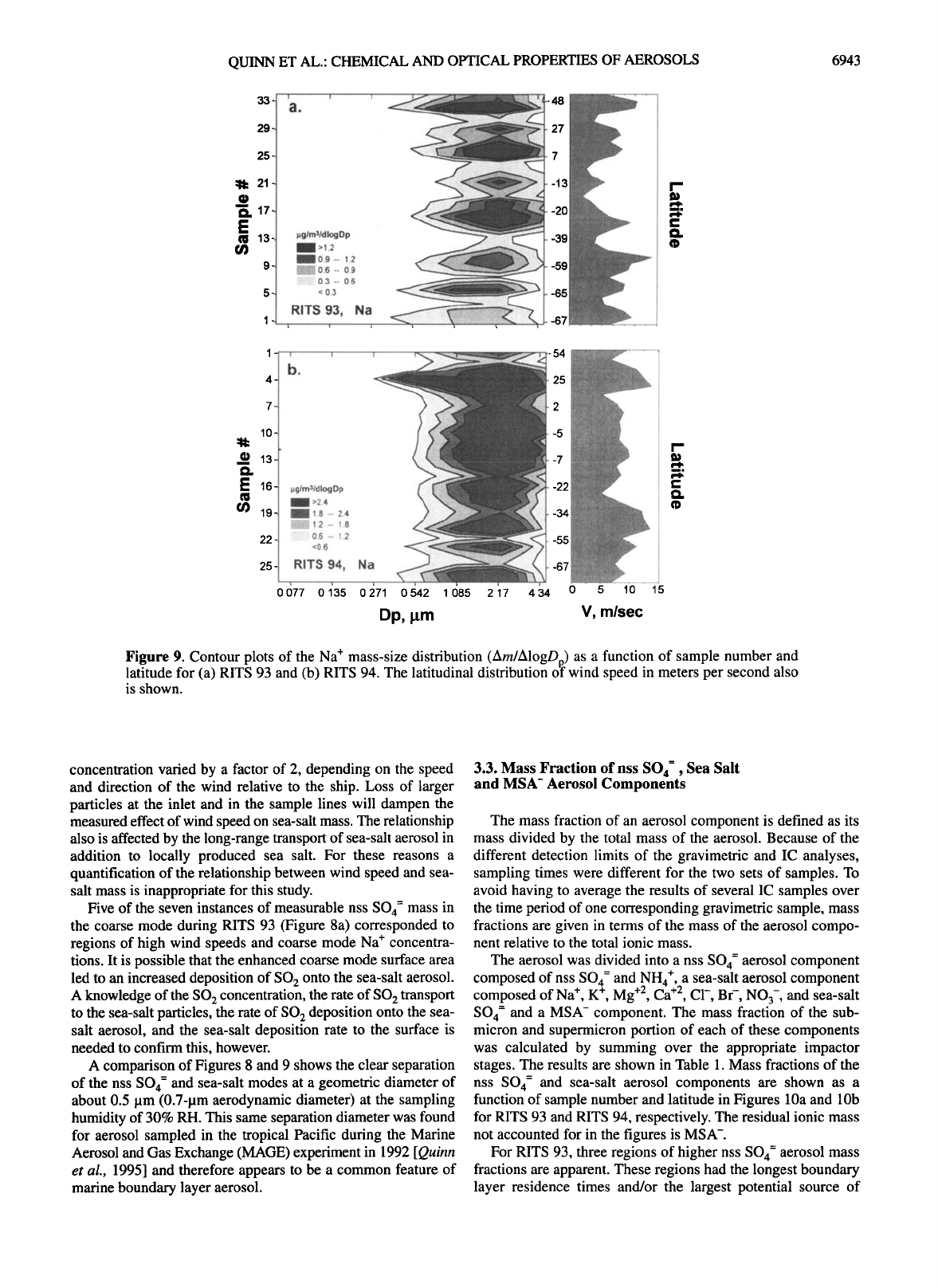
sea-salt aerosols (Figure 9a). A similar but less strongly corre-
lated pattern was observed for RITS 94 (Figure 9b). The
relationship between local wind speed and sea-salt aerosol mass
QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6943
33-
29-
25-
•1• 21-
O. 17-
E
{• 13-
9-
5-
1-
, I
p. glm31dlogDp
• >1.2
• 09 -- 1.2
•-- d-:•i ...... o.6 -- o.9
iiii•ii::!i!11iiii::ii.:: 0.3 -- 0.6
< 0.3
RITS 93, Na,
!
............... iiii•iii•:i,•,"" "-:: '"•
!i -39
¾•.:•i:• ......
- -155
- -67
10
__.• !3
• 16
03 19
22
25
b.
i•gim31d log Dp
• >2.4
• 1.8 -- 2.4
....... '"';;'"'""-'-'"•d;i 1.2 -- 1.8
...........
::::::::::::::::::::: 0.6 1.2
:•'•:•'•:•:F': --
<0.6
RITS 94, Na
0.077 0.135 0.27!
Dp, pm
.•.• ß , ., ...,.•
%:' ..... '":,:•!:!iii!ii!i!;;--":":":•!i 2 ":•i'-,:' ..-
: -,..-..;-•..:..:.-, ....::::...:. , '--<:'
' ,:-::-•'--'-';..::::•:•':" '. -' ' i
: '.,"'i':'"'--'gi:% ...... •:?:.• -5
..... . :"-.':-"-'T'"'"":'"'""'" ':'•: .............
:-.;: .--;..i•:....-j•j;•ii?::•ii;i;::i•,:• ....... ;::;?.!.../....½•4;-: -22
..... ..........
0.542 1.085 2.!7 4.34 0 5 10
V, rnlsec
15
Figure 9. Contour plots of the Na + mass-size distribution (Am/AlogDp) as a function of sample number and
latitude for (a) RITS 93 and (b) RITS 94. The latitudinal distribution o'f wind speed in meters per second also
is shown.
concentration varied by a factor of 2, depending on the speed
and direction of the wind relative to the ship. Loss of larger
particles at the inlet and in the sample lines will dampen the
measured effect of wind speed on sea-salt mass. The relationship
also is affected by the long-range transport of sea-salt aerosol in
addition to locally produced sea salt. For these reasons a
quantification of the relationship between wind speed and sea-
salt mass is inappropriate for this study.
Five of the seven instances of measurable nss SO4 = mass in
the coarse mode during RITS 93 (Figure 8a) corresponded to
regions of high wind speeds and coarse mode Na + concentra-
tions. It is possible that the enhanced coarse mode surface area
led to an increased deposition of SO 2 onto the sea-salt aerosol.
A knowledge of the SO 2 concentration, the rate of SO 2 transport
to the sea-salt particles, the rate of SO 2 deposition onto the sea-
salt aerosol, and the sea-salt deposition rate to the surface is
needed to confirm this, however.
A comparison of Figures 8 and 9 shows the clear separation
of the nss SO4 = and sea-salt modes at a geometric diameter of
about 0.5 pm (0.7-pm aerodynamic diameter) at the sampling
humidity of 30% RH. This same separation diameter was found
for aerosol sampled in the tropical Pacific during the Marine
Aerosol and Gas Exchange (MAGE) experiment in 1992 [Quinn
et al., 1995] and therefore appears to be a common feature of
marine boundary layer aerosol.
3.3. Mass Fraction of nss SO4 = , Sea Salt
and MSA- Aerosol Components
The mass fraction of an aerosol component is defined as its
mass divided by the total mass of the aerosol. Because of the
different detection limits of the gravimetric and IC analyses,
sampling times were different for the two sets of samples. To
avoid having to average the results of several IC samples over
the time period of one corresponding gravimetric sample, mass
fractions are given in terms of the mass of the aerosol compo-
nent relative to the total ionic mass.
The aerosol was divided into a nss SO4 = aerosol component
composed of nss SO4 = and NH4 +, a sea-salt aerosol component
+2 +2
composed of Na +, K +, Mg , Ca , CI-, Br-, NO3- , and sea-salt
SO4 = and a MSA- component. The mass fraction of the sub-
micron and supermicron portion of each of these components
was calculated by summing over the appropriate impactor
stages. The results are shown in Table 1. Mass fractions of the
nss SO 4- and sea-salt aerosol components are shown as a
function of sample number and latitude in Figures 10a and 10b
for RITS 93 and RITS 94, respectively. The residual ionic mass
not accounted for in the figures is MS A-.
For RITS 93, three regions of higher nss SO4 = aerosol mass
fractions are apparent. These regions had the longest boundary
layer residence times and/or the largest potential source of

6944 QUINN ET AL.' CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
Table 1. Percent Mass Fractions of Ionic Aerosol Components Relative to Measured Total or
Submicron Ionic Mass
RITS 93 RITS 94
Standard Standard
Component a Average Deviation Range Average Deviation Range
Percent Mass Fraction of Total Measured Ionic Mass
Super-gm sea salt 81 9 58-93 82 5.8 69-92
Sub-tam sea salt 10 7 1.4-28 8.3 4.6 2.7-19
Sub-tam nss SO4 = 7 8.3 0.2-34 6 3.7 0.9-13
Super-tam nss SO4 = 1.3 1 0.2-3.8 0.8 1 0.1-5.5
Sub-tam MSA- 0.39 0.48 0.08-2.7 1.0 1.6 0.03-6.0
Super-tam MSA- 0.24 0.37 0.03-2.0 0.40 0.29 0.03-1.1
Percent Mass Fraction of Submicron Measured Ionic Mass
Sub-tam sea salt 55 79 10-98 54 22 21-91
Sub-tam nss SO4 = 36 28 2-90 39 23 6-76
Sub-pm MSA- 2.1 1.7 0.31-10 5.5 6.6 0.37-24
Percent mass fractions are defined as the mass of the aerosol component divided by the measured
ionic mass.
aThe nss SO4 = aerosol component includes the associated NH4 + mass. The sea-salt aerosol
component includes Na +, K +, Mg +2, Ca +2, CI-, Br-, NO3-, and sea-salt SO4 =.
sulfate aerosol. From 27øN to 38øN where, according to number
and nss SO4 = concentrations, continentally influenced aerosol
had been advected in a nonprecipitating air mass from North
America, the mass fraction of nss SO4 = aerosol was the highest
measured, ranging from 23 to 37%. In the tropics, between 19 øS
and 2 øN, where boundary layer residence times were the longest
I• su•mSS •..•.•.*: ........... :::.•::::::.•::::::::.'.: ..................................... :" ............ • .................. =='
10 t• supumSS ................................... '•"• ............................................................... ß :':.-'•. -5
1•1 ...... ,,..x•x,•..•....,.,, .....: ........... ,..... ,...,,...., ..•... ::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: 1
• :::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: •
::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: •
:::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: 1
..,:.:::.:: =========================================================================================================================== ......
: :. :::•i:•:i:•:::•.}:•:•:•:..`•:•:•:g•:•:::::::.`.5.`.•:•.`.[•<:•:..`.i:.•..•:•:[:•:E:5:•:•:::5:[:•:[•:i•:[:{:;•:•:•:•:•:•:;•:•:s::•::` •l -67
::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: •
::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: •
0 20 40 60 80 100
Percent of total ionic mass
Figure 10. Mass fractions of the submicron nss SO4 =, super-
micron nss SO4 =, submicron sea-salt, and supermicron sea-salt
aerosol components as a function of sample number and latitude
for (a) RITS 93 and (b) RITS 94. Mass fractions are defined as
the mass of the aerosol component divided by the total ionic
mass as measured by ion chromatography. Residual ionic mass
not accounted for by these aerosol components is MSA-.
encountered (up to 7 days) and atmospheric DMS concentrations
were about 100 parts per trillion by volume (pptv), the mass
fraction was 5 to 20%. Values up to 10% occurred between
65øS and 67øS where atmospheric DMS concentrations of 200
pptv were among the highest measured along the cruise track.
Elsewhere, marine boundary layer residence times were less
than 5 days and DMS concentrations were less than 100 pptv.
A similar pattern is seen for RITS 94. Mass fractions of nss
SO4 = aerosol of 10% were observed between 65øS and 67øS.
This cruise reached the high southern latitudes during the austral
summer when ocean productivity in this region is at its maxi-
mum. The atmospheric DMS concentrations of up to 650 pptv
were the highest measured during both cruises. The higher mass
fractions in the tropics also were repeated during RITS 94 where
the values ranged from 8 to 16%. Mass fractions were not
enhanced between 20øN and 40øN during RITS 94, however, as
the sampled air had a northwest trajectory and had spent several
days over the ocean before reaching the ship.
3.4. Comparison of Measured and
Calculated Light Scattering
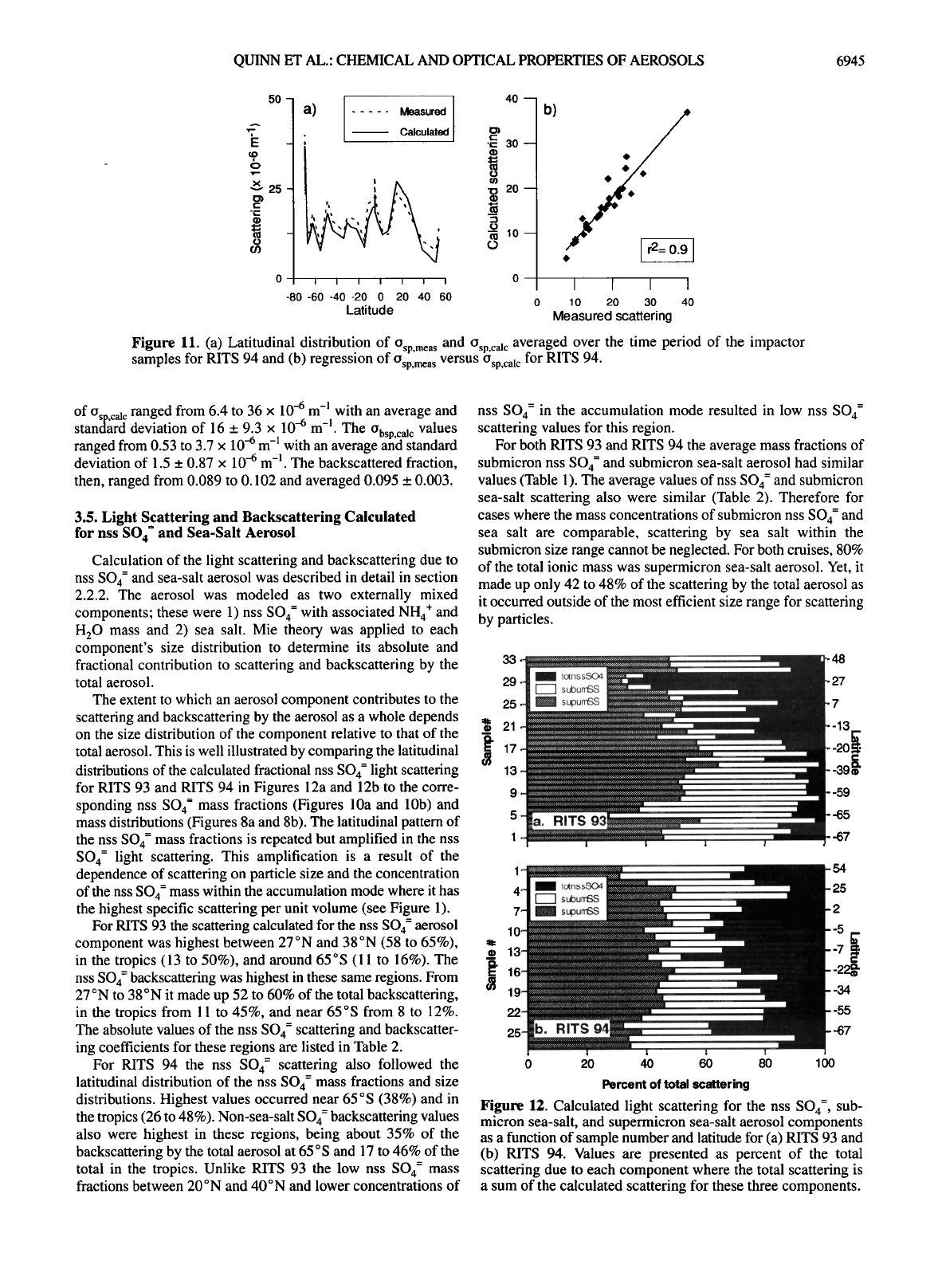
For RITS 94, (Jsp,meas ranged from 7.8 to 40 x 10 -6 m -1 with
an average and standard deviation of 18 + 6.8 x 10-6 m -1 (Figure
1 la). Values of O•p,c• c were similar, ranging from 4.3 to 37 x
10-6 m -I with an average and standard deviation of 16 _+ 6.7 x
10-6 m -1. A regression of the measured versus the calculated
values yields an r 2 value of 0.9 (Figure 11 b). This agreement is
within the uncertainty of the measurement and modeling
methods and indicates that the Mie model is able to parameterize
accurately the chemical and scattering characteristics of the total
aerosol. The Obsp,calc values ranged from 0.41 to 3.4 x 10-6 m -1
with an average and standard deviation of 1.5 + 0.62 x 10-6 m -].
The backscattered fraction based on O sp,cal c and Obsp,calc ranged
from 0.088 to 0.102 and averaged 0.095 + 0.004.
For RITS 93, Osp,mea s values were incorrect as a result of drift
in the sample and dilution airflows within the nephelometer.
Based on the agreement between the measured and calculated
values for the RITS 94 data set, o,p c•c instead of o,p meas values
were used in all further RITS 93 d•ta analyses. RIT• 93 values
QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6945
50-
•x 25-
-• _
a)
..... Measured
Calculated
I I I I I I I
-80-60-40-20 0 20 40 60
Latitude
40--
• 10--
o
b)
0 10 20 30 40
Measured scattering
Figure 11. (a) Latitudinal distribution of Osp,mea s and Osp,cal c averaged over the time period of the impactor
samples for RITS 94 and (b) regression of Osp,mea s versus Osp,cal c for RITS 94.
of Osp,cal c ranged from 6.4 to 36 x 10 -6 m -• with an average and
standard deviation of 16 _+ 9.3 x 10 -6 m -]. The Obsp,calc values
ranged from 0.53 to 3.7 x 10 -6 m -! with an average and standard
deviation of 1.5 _+ 0.87 x 10 -6 m -]. The backscattered fraction,
then, ranged from 0.089 to 0.102 and averaged 0.095 _+ 0.003.
3.5. Light Scattering and Backscattering Calculated
for nss SO4 = and Sea-Salt Aerosol
Calculation of the light scattering and backscattering due to
nss SO4 = and sea-salt aerosol was described in detail in section
2.2.2. The aerosol was modeled as two externally mixed
components; these were 1) nss SO4 = with associated NH4 + and
H20 mass and 2) sea salt. Mie theory was applied to each
component's size distribution to determine its absolute and
fractional contribution to scattering and backscattering by the
total aerosol.
The extent to which an aerosol component contributes to the
scattering and backscattering by the aerosol as a whole depends
on the size distribution of the component relative to that of the
total aerosol. This is well illustrated by comparing the latitudinal
distributions of the calculated fractional riss SO4 = light scattering
for RITS 93 and RITS 94 in Figures 12a and 12b to the corre-
sponding nss SO4 = mass fractions (Figures 10a and 10b) and
mass distributions (Figures 8a and 8b). The latitudinal pattern of
the nss SO4 = mass fractions is repeated but amplified in the nss
SO4 = light scattering. This amplification is a result of the
dependence of scattering on particle size and the concentration
of the nss SO4 = mass within the accumulation mode where it has
the highest specific scattering per unit volume (see Figure 1).
For RITS 93 the scattering calculated for the nss SO4 = aerosol
component was highest between 27øN and 38øN (58 to 65%),
in the tropics (13 to 50%), and around 65øS (11 to 16%). The
nss SO4 = backscattering was highest in these same regions. From
27øN to 38øN it made up 52 to 60% of the total backscattering,
in the tropics from 11 to 45%, and near 65øS from 8 to 12%.
The absolute values of the nss SO4 = scattering and backscatter-
ing coefficients for these regions are listed in Table 2.
For RITS 94 the nss SO4 = scattering also followed the
latitudinal distribution of the nss SO4 = mass fractions and size
distributions. Highest values occurred near 65øS (38%) and in
the tropics (26 to 48%). Non-sea-salt SO4 = backscattering values
also were highest in these regions, being about 35% of the
backscattering by the total aerosol at 65øS and 17 to 46% of the
total in the tropics. Unlike RITS 93 the low nss SO4 = mass
fractions between 20 øN and 40øN and lower concentrations of
nss SO4 = in the accumulation mode resulted in low nss SO4 =
scattering values for this region.
For both RITS 93 and RITS 94 the average mass fractions of
submicron nss SO4 = and submicron sea-salt aerosol had similar
values (Table 1 ). The average values of nss SO4 = and submicron
sea-salt scattering also were similar (Table 2). Therefore for
cases where the mass concentrations of submicron nss SO4 = and
sea salt are comparable, scattering by sea salt within the
submicron size range cannot be neglected. For both cruises, 80%
of the total ionic mass was supermicron sea-salt aerosol. Yet, it
made up only 42 to 48% of the scattering by the total aerosol as
it occurred outside of the most efficient size range for scattering
by particles.
33 .<- ...... ß ............................. • ......................................... ,, .... . 48
:.::.::.:..•:.:...:.:....:..,:..,.•xxxx,-,,.,,.,,,vx• i
29 ß • t•nss•
......... 27
:: • s•u• ............
25 : • s•u• ................................ 7
• 21 ........................................................................... 13
• ...... :::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::: ............................
17 .......................... ;•:•.•.•2•7•`;}•<:::•7``;;7•`•7•;•>•.•``:`2•``•;:`•J<•::•.•;•:;•`;•2•` -20 •.
9 •``•``*•``•.`*•```•``•`•```•<<*•``•.•`•.•>•:•.. -59
:::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::
:+xx.x•,x,x,:.:.xx.xx.:.:.x.>x,•x,x.:.:.x.:.>>:.:.x.x+x,x.x,x.:,,:..
•>>:.>:.:.:.:.•.:.:.:.:.>x.•.>>•.:.•.•.:.>m.:•:.:.:.•.:.•.•.:.•.•.:•>x`•:.:.x.•.•.:.:.x.:..
5 •a. RITS 93 ................................................................................... -65
.x.>:.x.:•,x.x<<,x•<,,x.:.:•<<.:.:..,:.:<,,:.x.:.x<•,xx+•,x•xx,>x•,x•x,:,xq
1 ............................................................................................................. •7
4 I totnssSO4 ::::::::::::::::::::::::::::
:•:•:..,.?/.,..:•,<:•:•:•:?/::.x;:..,.•:•$?/:•:
ß 1= ::::: .•:::•::•:: ,x3 •:: •:•: :: :.<:•:: •<::: •:::: :: :s:::::: •::.,..!: :: 3: ::::::::::::::::::::::::: :::::: •::::: :: s :::• .,.:: :::::::.:.
(j• :!:.•:`.``3x.`.•3:i:•3.``3•.``.:•:•!.`..`q•:!:•3::•:..`.::•:•:.•:•:•:;:•.x.•:3:•:•:.:..•:35:•:3•!8?.`..•:!:.``i•:!:•:3•
:::::...:.:.:.:.:.:.:.:.:.:.:.:.:.:.:.:.-...:.:.....: x... =================================================
25 ::i:::b. RITS 94
:•::...•.:.:.:`:.:.:.x•.:.:.:.:.`•:.`.•.:.:.:.:...:.:.•...:.•.:.•...•.:.:+:.:•::::::::.•:::::•
:::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::::
0 20 40 60 80 lOO
54
25
2
-5 g.
-7 •.
-22•
-34
-55
-67
Percent of total scattering
Figure 12. Calculated light scattering for the nss SO4 =, sub-
micron sea-salt, and supermicron sea-salt aerosol components
as a function of sample number and latitude for (a) RITS 93 and
(b) RITS 94. Values are presented as percent of the total
scattering due to each component where the total scattering is
a sum of the calculated scattering for these three components.

6946 QUINN ET AL.' CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
Table 2. Scattering and Backscattering Coefficients Calculated for nss SO4 =, Submicron Sea
Salt, and Supermicron Sea-Salt Aerosol Components for Latitudinal Regions Where nss SO 4-
Had the Largest Contribution to Scattering by Total Aerosol
NH Midlatitudes Tropics
27øN to 38øN 20øS to 20øN
Coefficient,
x 10 -6 m -1 RITS 93 RITS 94 RITS 93 RITS 94 RITS 93 RITS 94
S H Hi gh Latitudes
65 øS to 67øS
11-18 1.8-2.4 1.4-9.4 2.9-9.3 1.3-2.1 1.7-5.6
IJsp,SO4,aer
Osp,sub,seasalt 0.43-2.5 2.9-8.6 0.72-11 1.8-11 2.5-13 2.3-18
Osp,sup,seasalt 5.2-10 3.2-11 2.6-19 3.8-13 2.3-6.7 3.7-13
Obsp,SO4,ae r 0.89--1.4 0.12-0.16 0.12--0.62 0.22-0.83 0.1-0.13 0.33--0.39
Obsp,sub,seasal t 0.1-0.23 0.23-0.71 0.10-0.95 0.16-0.85 0.21-1.0 0.19-1.5
Obsp,sup,seasal t 0.57-1.1 0.35-1.2 0.28-2.1 0.41-1.5 0.27-1.1 0.41-1.5
NH, northern hemisphere; SH, southern hemisphere.
3.6. Mass Scattering Efficiencies for the nss SO4--
and Sea-Salt Aerosol Components
Mass scattering efficiencies for individual aerosol com-
ponents can be estimated from a multiple linear regression of the
mass concentration of each aerosol component against the
scattering coefficient for the whole aerosol. For the RITS 93 and
RITS 94 data sets a regression of the following form, including
only the major aerosol components, was used to obtain weighted
averages of the scattering efficiencies
: {z tn
(Jsp,calc {Zsp,SO4,ion mso4,ion + sp,sub,seasalt sub,seasalt
+ sp,sup,seasalt sup,seasalt
•z m
(4)
The mass scattering efficiency of nss SO4 = ({Zsp,SO4,ion) is given
in terms of the unit mass of nss SO 4- ion as it is the ion concen-
tration or column burden that is predicted by chemical transport
models [e.g., Langner and Rodhe, 1991]. Values of mso4,ion
include both the submicron and supermicron size fractions since
the majority of the nss SO 4- mass (>75%) occurred in the
submicron size range and this is the size range expected to have
the most significant effect on scattering by the total aerosol.
Unlike the nss SO4 = aerosol component, the majority of the
sea-salt aerosol mass was in the supermicron size fraction which
will not have a large influence on scattering by the total aerosol
unless it overwhelms the total particle mass concentration. To
consider the more relevant submicron fraction, the sea-salt
component was divided into submicron and supermicron size
ranges resulting in a submicron mass scattering efficiency
({gsp,sub,seasalt) and a supermicron mass scattering efficiency
({gsp,sup,seasalt). Mass backscattering efficiencies for the aerosol
components were calculated by substituting the calculated
backscattering coefficient, IJbsp,calc, for the whole aerosol into
(4).
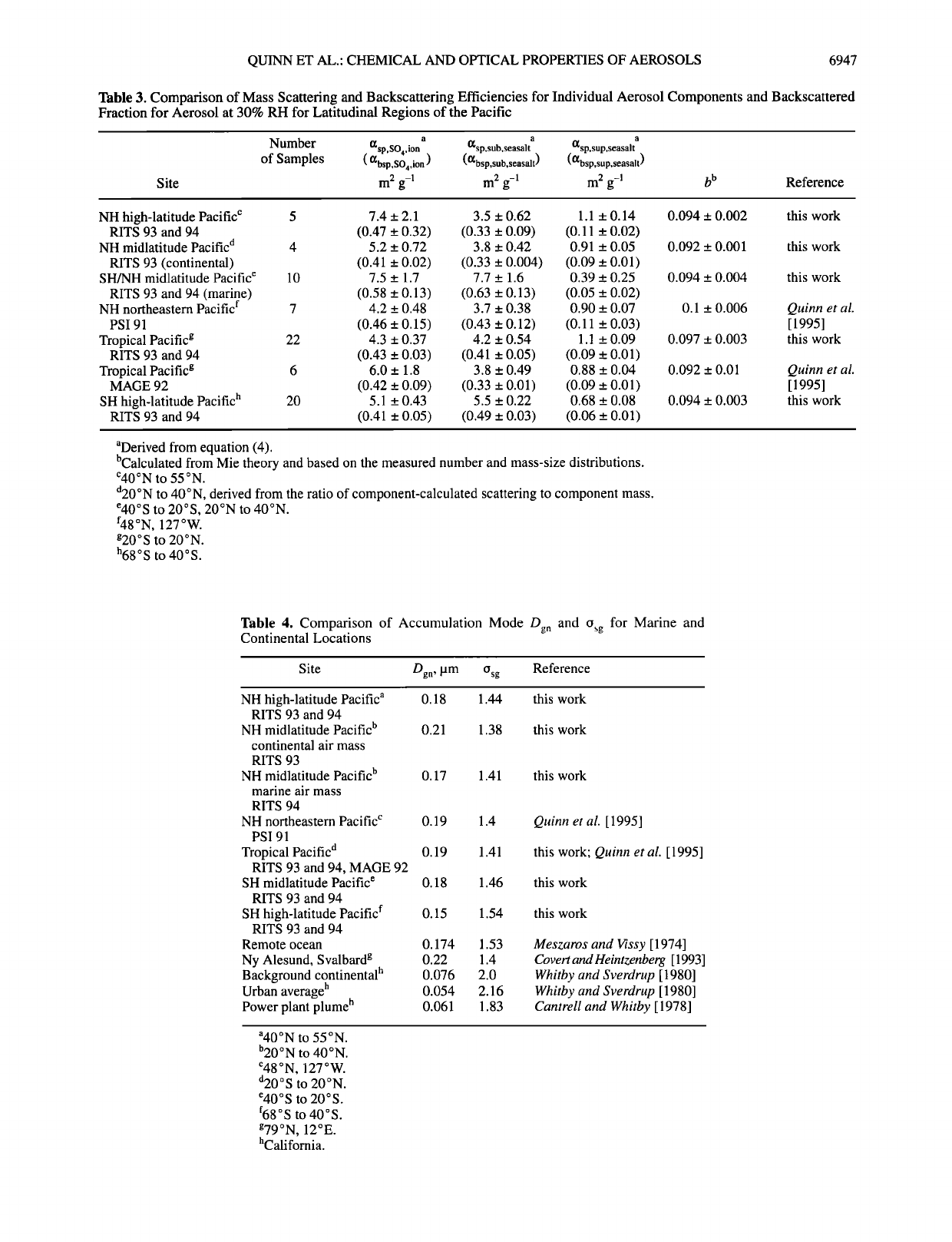
The calculated mass scattering and backscattering efficiencies
averaged over latitudinal regions of the Pacific from RITS 93
and RITS 94 are shown in Table 3. For RITS 93 the weighted
average and standard error from the regression of {gsp,SO4,ion for
all samples were 5.1 + 0.21 m 2 g-• and for RITS 94, 3.8 + 0.35
m 2 g-•. Values were relatively constant over all latitudinal
regions with weighted averages ranging from 4.3 to 7.5 m 2 g-•.
The stability of the values is a reflection of the narrow range of
Dg n and Osg measured throughout both cruises (see Table 4).
More variability in {gsp,SO4,ion is expected in instances where Dg n
and Osg for the nss SO4 = aerosol component vary in space or
time (see, for example, Zhang et al. [1994]). The largest values
were observed in the northern hemisphere high latitudes (40 øN
to 55 øN) and in marine air masses in the southern and northern
hemisphere midlatitudes (40øS to 20øS and 20øN to 40øN).
Lowest values were found in the tropics between 20øS and
20øN.
The average and standard deviation of {gbsp,SO4,ion was about
an order of magnitude lower and displayed less variability than
the scattering efficiency values. The weighted average and
standard error of all RITS 93 samples were 0.43 + 0.02 m 2 g-•
and for RITS 94, 0.40 + 0.03 m 2 g-•. Weighted averages ranged
from 0.41 to 0.58 m 2 g-• over all latitudinal regions. The lower
degree of variability in the backscattering efficiencies compared
to the scattering efficiencies is a result of a weaker dependence
of Obs p than Osp on particle size for diameters between 0.2 and
1.0 pm [Marshall, 1994].
The weighted average and standard error of {gsp,sub,seasalt for
RITS 93 were 5.0 + 0.27 m 2 g-• and for RITS 94, 4.9 +
0.24 m 2 g-•. Values of {gbsp,sub,seasal t for RITS 93 averaged 0.47
2 1
_+ 0.03 m g- and for RITS 94 averaged 0.42 _+ 0.02 m 2 g-•.
The equivalent values for {gsp,SO4,ion and {gsp,sub,seasalt as well as
{gbsp,SO4,ion and {gbsp,sub,seasal t indicate that if the submicron masses
of sea salt and nss SO4 = are comparable, the contribution by
submicron sea salt to scattering and backscattering by the
aerosol as a whole cannot be neglected given that the submicron
sea salt and nss SO4 = aerosol have similar lifetimes in the
atmosphere.
Mass scattering and backscattering efficiencies for the
supermicron sea-salt component were appreciably lower,
however, as it is in a size range where particle scattering in the
visible is less efficient per unit volume. RITS 93 and RITS 94
2 1
values of {ZSlP,SUp,seasal t averaged 0.88 _+ 0.05 m g- and 0.92 _+
0.07 m 2 g-, respectively. RITS 93 and RITS 94 values of
{gbsp,sup,seasal t averaged 0.08 + 0.01 m 2 g-• and 0.09 + 0.01 m 2
respectively. The lack of variability in the scattering and
backscattering efficiencies of the sea-salt aerosol suggests that
QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6947
Table 3. Comparison of Mass Scattering and Backscattering Efficiencies for Individual Aerosol Components and Backscattered
Fraction for Aerosol at 30% RH for Latitudinal Regions of the Pacific
Number i•sp,so4,ion a i•sp,sub,seasalt a i•sp,sup,seasalt a
of Samples ( {gbsp,SO4,io n ) ({gbsp,sub,seasalt) ({gbsp,sup,seasalt)
Site m 2 g-• m 2 g-• m 2 g-• b b Reference
NH high-latitude Pacific c 5 7.4 _+ 2.1 3.5 _+ 0.62 1.1 _+ 0.14 0.094 _+ 0.002 this work
RITS 93 and 94 (0.47 _+ 0.32) (0.33 _+ 0.09) (0.11 _+ 0.02)
NH midlatitude Pacific d 4 5.2 _+ 0.72 3.8 _+ 0.42 0.91 _+ 0.05 0.092 _+ 0.001 this work
RITS 93 (continental) (0.41 + 0.02) (0.33 + 0.004) (0.09 + 0.01)
SH/NH midlatitude Pacific e 10 7.5 + 1.7 7.7 + 1.6 0.39 + 0.25 0.094 +_ 0.004 this work
RITS 93 and 94 (marine) (0.58 _+ 0.13) (0.63 _+ 0.13) (0.05 _+ 0.02)
NH northeastern Pacific f 7 4.2 + 0.48 3.7 + 0.38 0.90 + 0.07 0.1 + 0.006 Quinn et al.
PSI91 (0.46_+0.15) (0.43_+0.12) (0.11 _+0.03) [1995]
Tropical Pacific g 22 4.3 _+ 0.37 4.2 _+ 0.54 1.1 +_ 0.09 0.097 _+ 0.003 this work
RITS 93 and 94 (0.43 _+ 0.03) (0.41 _+ 0.05) (0.09 _+ 0.01)
Tropical Pacific g 6 6.0 _+ 1.8 3.8 _+ 0.49 0.88 _+ 0.04 0.092 _+ 0.01 Quinn et al.
MAGE 92 (0.42 _+ 0.09) (0.33 _+ 0.01) (0.09 _+ 0.01) [ 1995]
SH high-latitude Pacific h 20 5.1 _+ 0.43 5.5 _+ 0.22 0.68 _+ 0.08 0.094 _+ 0.003 this work
RITS 93 and 94 (0.41 _+ 0.05) (0.49 _+ 0.03) (0.06 _+ 0.01)
aDerived from equation (4).
bCalculated from Mie theory and based on the measured number and mass-size distributions.
c40øN to 55øN.
d20øN to 40øN, derived from the ratio of component-calculated scattering to component mass.
e40øS to 20øS, 20øN to 40øN.
f48øN, 127øW.
g20øS to 20øN.
h68 o S to 40 ø S.
Table 4. Comparison of Accumulation Mode Dg n and Osg for Marine and
Continental Locations
Site Dgn, t am (Jsg Reference
NH high-latitude Pacific a 0.18 1.44
RITS 93 and 94
NH midlatitude Pacific b 0.21 1.38
continental air mass
RITS 93
NH midlatitude Pacificb 0.17 1.41
marine air mass
RITS 94
NH northeastern Pacific c 0.19 1.4
PSI 91
Tropical Pacific d 0.19 1.41
RITS 93 and 94, MAGE 92
SH midlatitude Pacific e 0.18 1.46
RITS 93 and 94
SH high-latitude Pacific f 0.15 1.54
RITS 93 and 94
Remote ocean 0.174 1.53
Ny Alesund, Svalbard g 0.22 1.4
Background continental h 0.076 2.0
Urban average h 0.054 2.16
Power plant plume h 0.061 1.83
this work
this work
this work
Quinn et al. [1995]
this work; Quinn et al. [1995]
this work
this work
Meszaros and Vissy [1974]
Covert and Heintzenberg [ 1993]
Whitby and Sverdrup [ 1980]
Whitby and Sverdrup [ 1980]
Canttell and Whitby [1978]
a40øN to 55øN.
b20øN tO 40øN.
c48 øN, 127 øW.
d20øS tO 20øN.
%0 o S to 20 ø S.
f68 o S to 40 ø S.
g79 øN, 12øE.
hCalifornia.

6948 QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
the sea salt was sampled close to its source; its size distribution
did not vary significantly from sample to sample (see Figure 6);
and it did not undergo much alteration via gas phase absorption,
cloud processing, or removal prior to being sampled.
4. Comparison of Climate Relevant Parameters
Derived From RITS 93 and RITS 94 With Pre-
viously Reported Marine Values
Determining AF• for tropospheric aerosols requires knowing
how perturbations to the natural aerosol system through the
addition of anthropogenic aerosol changes the chemical,
physical, and radiative properties of the natural aerosol. These
properties include the number distribution of the aerosol as a
whole, the amount of light that is scattered in the upward
direction by the aerosol as a whole, and the mass scattering and
backscattering efficiencies of each aerosol component (natural
and anthropogenic) present. Several recent cruises in the Pacific
Ocean have allowed for a compilation of these quantities for
aerosol in the marine atmosphere.
Geometric number mean diameters, Dgn, and standard
deviations, Osg ' for different regions of the Pacific and a selection
of continental locations are listed in Table 4. Values of Dg n and
Osg from marine air masses cover a narrow range from 0.15 to
0.19 and 1.4 to 1.54, respectively. Continentally influenced
marine air masses (northern hemisphere midlatitude Pacific
during RITS 93) and remote continental air masses (Ny
Alesund) have slightly larger values of Dg n (0.21 and 0.22) but
similar standard deviations (1.38 and 1.4). These values of Dg n
indicate that the sampled marine and remote continental aerosol
had undergone some degree of aging and relatively rapid growth
to the Aitken mode since production. The narrow values of Osg
suggest, however, that the number and variability of growth
mechanisms to the accumulation size was limited. Nonremote
continental air masses from California have much smaller
geometric number mean diameters of 0.054 to 0.076 but larger
standard deviations of about 2.0. These values are indicative of
an aerosol that is close to its source. The growth that has
occurred, however, has been a result of a number of mecha-
nisms.
Values of the backscattered fraction, b, for different regions
of the Pacific derived from measurements during RITS 93, RITS
94, MAGE 92, and the Pacific Sulfur Stratus Investigation (PSI
91) are compared in Table 3. Values are consistent for the four
cruises, coveting a narrow range from 0.092 to 0.1. This
consistency is a result of the relative stability of the shape (but
not necessarily magnitude) of the accumulation mode and
suggests that only a limited number of aerosol production and
growth mechanisms occur in the marine atmosphere. The
relatively narrow range of values observed for Dg n and Osg in
marine air masses confirms this stability.
Mass scattering and backscattering efficiencies for nss SO4 =,
submicron sea salt, and supermicron sea salt at 30% RH for
these four cruises also are shown in Table 3. Values for all of the
cruises were estimated from a multiple linear regression of the
Mie-calculated scattering by the aerosol as a whole against the
aerosol component masses (equation (4)). The mass scattering
efficiencies for all three components are consistent among the
four cruises. Average values of ttsp,S% ion range from 4.2 to
2 -1 "2 -1
7.5 m g , •sp,sub,seas•lt fr•om 3.5 to 7.7 m g , and •sp,sup, seasalt
from 0.68 to 1.1 m g-'. Mass backscattering efficiencies of
{gbsp,SO4,ion range from 0.41 to 0.58 m 2 g-l, 0•bspsub seasalt from
0.33 to 0.63 m 2 g-i, and •bsp,sup,seasalt from 0.05 (O 6.11 m 2
Many values of mass scattering efficiencies for sulfate
aerosol have been reported for continental regions. These
include 2.0 to 6.0 m 2 g-1 from the Grand Canyon [Zhang et al.,
1994; Anderson et al., 1994], 8 m 2 g-1 from Denver [Sloane et
al., 1991], 5 m 2 g-1 from southern Sweden [Waggoner et al.,
1976], 11 m 2 g-1 from the Canadian Arctic [Barrie and Hoff,
1985], and 12 m 2 g-1 from the northeastern United States [ten
Brink et al., 1987]. A direct comparison of these numbers with
each other and with the marine values reported here is difficult,
however, because of the lack of a standard definition of the
sulfate mass scattering efficiency.
The values of sulfate mass scattering efficiencies from this
work are weighted averages based on Mie-calculated scattering
by the aerosol as a whole and the mass concentration of the
aerosol components. This calculation preserves the size resolu-
tion of the measured number distribution and takes into account
the nss SO4 = size distribution. Zhang et al. [1994] use Mie-
calculated ammonium sulfate scattering based on measured
chemical mass distributions to estimate the sulfate mass
scattering efficiency. Values from this work and from Zhang et
al. [ 1994] assume that the aerosol is an external mixture. Sloane
et al. [1991] derive sulfate scattering efficiencies in a similar
manner but with models which assume either an externally or an
internally mixed aerosol. These results indicate that the assump-
tion of an external mixture yields scattering efficiencies 15 to
30% lower than if an internal mixture is assumed. In each of
these methods it is assumed that only those chemical species that
were analytically measured contributed to scattering by the total
aerosol. If other species which also contribute to scattering are
present, then the sulfate mass scattering efficiencies will be
artificially high.
Values of sulfate mass scattering efficiencies reported by ten
Brink et al. [1987], Waggoner et al. [1976], and Barrie and Hoff
[ 1985] are based on the measured light scattering by the total
aerosol divided by the measured mass concentration of the nss
SO4 = ion. This method does not take into account the size
distribution of the sulfate aerosol component. In addition, the
sulfate scattering efficiencies will be overestimated if aerosol
components other than nss SO4 = are present and are contributing
to measured scattering by the total aerosol.
Reported values of sulfate mass scattering efficiencies also
will be affected by the spectral response of the nephelometer
light source, light filter, and detector phototube. Differences in
this response are difficult to quantify, however, as they are a
function of the number distribution which can vary greatly
between different types of air masses. Ruby and Waggoner
[1981] have estimated, for example, that differences in the
spectral response of the Meteorology Research, Inc. (MRI)
nephelometer models 1550 and 1560/90 can result in light
scattering coefficients that differ by 6 to 26% depending on the
size distribution.
Differences in the relative humidity of the scattering measure-
ment also can lead to variability in reported sulfate scattering
efficiencies. This effect is variable as it depends on the deliques-
cence properties of the nss SO4 = aerosol which, in turn, depend
on the degree of neutralization of the sulfate by ammonium.
Changes in ambient RH from 60 to 90% have been found to
affect sulfate scattering efficiencies by as much as a factor of 2
to 3 [Weiss et al., 1982; ten Brink et al., 1987; Zhang et al.,
1994]. As a result, scattering efficiencies will vary greatly if
derived from nephelometer measurements of a dried airstream
versus one at the ambient RH. One other factor affecting the
magnitude of the sulfate scattering efficiency is the size fraction

QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6949
of the nss SO4 = mass that is included in the calculation. If there
is a significant amount of supermicron nss SO4 = mass, using a
combined submicron and supermicron value will give a consid-
erably different value than using only a submicron value as the
two size ranges have different scattering characteristics.
On the basis of the above discussion it is difficult to deter-
mine how much of the variability in reported values of sulfate
scattering efficiencies is due to differences in aerosol properties
versus how much is due to differences in measurement and
calculation methods. For the purpose of modeling the climatic
effect of tropospheric aerosol on a global scale, a large database
of scattering and nss SO4 = mass distribution measurements is
needed to indicate regional and temporal variability in the
sulfate scattering efficiency. Such a database must rely on a
standardized method for estimating the sulfate scattering
efficiency so that variability due to methods is minimized.
Extending this standardization to the determination of the mass
scattering efficiency for all aerosol components that contribute
to scattering by the total aerosol would be very useful in
determining the roles of different aerosol types in the climatic
effect of tropospheric aerosol.
5. Conclusions
Presented here are data from two long latitudinal cruises in
the Pacific Ocean which allow for the determination of the effect
of air mass sources and synoptic scale meteorology on the
chemical, physical, and optical properties of individual aerosol
components. These empirical relationships are needed to assess
the perturbation of the radiative properties of the natural aerosol
system by the addition of anthropogenic aerosol and to incorpo-
rate the direct climate forcing by tropospheric aerosol into
global climate models.
Frequent occurrence of frontal passages and the resulting
subsidence of air from the free troposphere in the high latitudes
(>45 ø ) of the northern and southern hemispheres led to short
marine boundary layer residence times of less than 3 days.
Subsidence and transport also occurred on the edges of high-
pressure systems in the midlatitudes (20 ø to 40ø), yielding
variable boundary layer residence times of 1 to 5 days. A
tropical depression between midlatitude highs (20øS to 20øN)
resulted in long boundary layer residence times of up to 7 days.
Subsidence and short boundary layer residence times corre-
sponded to high Aitken mode number concentrations and low
accumulation mode number and nss SO4 = mass concentrations.
Alternatively, long boundary layer residence times allowed for
growth of the aerosol from the Aitken to the accumulation mode
size range and resulted in a less prominent Aitken mode and
high accumulation mode number and nss SO4 = mass concentra-
tions.
In all regions, high Na + concentrations corresponded to high
wind speeds, indicating the wind-driven production of sea-salt
aerosols. The nss SO4 = and Na + masses were clearly separated
into submicron and supermicron particle size fractions, respec-
tively, with a separation geometric diameter of 0.5 tam (0.7-tam
aerodynamic diameter) for the aerosol at 30% RH. As shown in
Figure 1, the minimum between the two modes for the marine
case requires an aerodynamic size cut ranging between 0.7 to 1.0
tam to separate the two modes.
Submicron nss SO4 = aerosol made up 35 to 40% of the
submicron ionic mass and 6% of the total ionic mass. Super-
micron nss SO4 = aerosol contributed only 1% to the total ionic
mass. Mass fractions of nss SO4 = aerosol were highest in regions
having the longest marine boundary layer residence times (the
tropics) or the largest potential sources of sulfate aerosol (>65 øS
where atmospheric DMS concentrations were high and the
northern hemisphere midlatitudes where air was advected from
North America). Mass fractions of submicron sea-salt aerosol
were similar to those of submicron nss SO4 =.
The latitudinal distribution of nss SO4-- light scattering
followed that of the nss SO4 = mass fraction but was amplified
due to the concentration of the sulfate aerosol in the accumula-
tion mode where the scattering coefficient per unit aerosol
volume is highest. Largest values were found in the high
southern latitudes (up to 38% of the scattering by the total
aerosol), in the tropics (up to 48%), and in the northern hemi-
sphere between 27øN and 38øN (up to 65%). Values of the
submicron sea-salt light scattering were similar to those of the
sulfate aerosol. Supermicron sea-salt aerosol made up 80% of
the total ionic mass but only contributed about 45% to the
scattering by the total aerosol due to the low scattering coeffi-
cient at visible wavelengths per unit aerosol volume in this size
range. In addition, scattering by supermicron sea-salt aerosols
will only be important at low altitudes as particles of this size
generally are confined to the lowest layer of the atmosphere due
to their large sedimentation velocities.
Mass scattering and backscattering efficiencies for the nss
SO4 = ion ranged from 4.3 to 7.5 and from 0.41 to 0.58 m 2 g-i,
respectively. Scattering and backscattering efficiencies for
submicron sea salt ranged from 3.5 to 7.7 and from 0.33 to 0.63
m 2 g-i, respectively. The similarity of the scattering efficiencies
for sulfate and submicron sea salt indicates that the contribution
by submicron sea salt to scattering by the total aerosol cannot be
neglected for cases where the mass concentrations of the two
components are similar. Mass scattering efficiencies of super-
micron sea salt were considerably lower.
A direct comparison of the mass scattering efficiencies for the
nss SO4 = ion presented here to those reported for more continen-
tal regions is difficult because of differences in the methods used
for the estimation. Variability in sulfate mass scattering efficien-
cies can result from natural variability in aerosol properties but
also can be due to the use of Mie-calculated scattering versus
total measured scattering, differences in the spectral responses
of the nephelometers used for the scattering measurement,
differences in the measurement RH, and the use of different size
fractions of the nss SO4 = ion. A standardized protocol for the
determination of sulfate mass scattering efficiencies is needed
to eliminate variability due to measurement methods. Given this
standardized method, a global database of sulfate scattering
efficiencies indicating the regional and temporal variability of
sulfate aerosol properties can be developed to incorporate the
direct effect of aerosols into climate models.
Values of the backscattered fraction, b, were comparable to
previously reported marine values ranging from 0.092 to 0.1.
This consistency is a result of stability in the shape of the
accumulation mode over different latitudinal and aerosol source
regions. The narrow range of values observed for Den (0.15 to
0.21 tam) and os• (1.38 to 1.54) confirms this stability.
The data presented here consider the mass fractions and light
scattering of two aerosol components, nss SO4 = and sea-salt
aerosol. To determine how anthropogenic perturbations to the
natural aerosol system change the radiative properties of the
aerosol, this analysis should be extended to all optically relevant
aerosol components, including nss SO4-, sea salt, organic
carbon, elemental carbon, and mineral aerosol.
Acknowledgments. We thank C. Zenker for analytical
assistance, the officers and crew of the Surveyor for their coopera-

6950 QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS
tion, and T. Anderson and R. Charlson for helpful comments. This
research was funded by the Ecology and Atmospheric Chemistry
Branch of the NASA Mission to Planet Earth Science Division and
by the Aerosol Component of the NOAA Climate and Global
Change Program. It is a contribution to the International Global
Atmospheric Chemistry (IGAC) Core Project of the International
Geosphere-Biosphere Programme (IGBP). This is NOAA PMEL
contribution 1645 and JISAO contribution 326.
References
Anderson, T. L., R. J. Charlson, W. H. White, and P. H. McMurry,
Comment on "Light scattering and cloud condensation nucleus
activity of sulfate aerosol measured over the Northeast Atlantic
Ocean" by D. A. Hegg et al., J. Geophys. Res., 99, 25,947-
25,949, 1994.
Andreae, M. O., The ocean as a source of atmospheric sulfur
compounds, in The Role of Air-Sea Exchange in Geochemical
Cycling, edited by P. Buat-Menard, pp. 331-362, D. Reidel,
Norwell, Mass., 1986.
Baron, P. A., Calibration and use of the aerodynamic particle sizer
(APS 3300), Aerosol Sci. Technol., 5, 55-67, 1986.
Barfie, L. A., and R. M. Hoff, Five years of air chemistry observa-
tions in the Canadian Arctic, Atmos. Environ., 19, 1995-2010,
1985.
Bates, T. S., J. D. Cline, R. H. Gammon, and S. R. Kelly-Hansen,
Regional and seasonal variations in the flux of oceanic
dimethylsulfide to the atmosphere, J. Geophys. Res., 92,
2930-2938, 1987.
Bates, T. S., K. C. Kelly, J. E. Johnson, and R. H. Gammon,
Regional and seasonal variations in the flux of oceanic carbon
monoxide to the atmosphere, J. Geophys. Res., 100, 23,093-
23,101, 1995.
Bemer, A., C. Lurzer, F. Pohl, O. Preining, and P. Wagner, The
size distribution of the urban aerosol in Vienna, Sci. Total
Environ., 13, 245-261, 1979.
Bodhaine, B. A., N. C. Ahlquist, and R. C. Schnell, Three-wave-
length nephelometer suitable for aircraft measurement of
background aerosol scattering coefficient, Atmos. Environ.,
25A, 2267-2276, 1991.
Bohren, C. F., and D. R. Huffman, Absorption and Scattering of
Light by Small Particles, John Wiley, New York, 1983.
Bray, W. H., Water vapor pressure control with aqueous solutions
of sulfuric acid, J. Mater., 5, 233-248, 1970.
Cantrell, B. K., and K. T. Whitby, Aerosol size distributions and
aerosol volume formation for a coal-fired power plant plume,
Atmos. Environ., 12, 323-333, 1978.
Clarke, A.D., Atmospheric nuclei in the Pacific midtroposphere:
Their nature, concentration, and evolution, J. Geophys. Res., 98,
20,633-20,647, 1993.
Covert, D. S., and J. Heintzenberg, Size distributions and chemical
properties of aerosol at Ny Alesund, Svalbard, Atmos. Environ.,
27A, 2989-2997, 1993.
Covert, D. S., V. N. Kapustin, T. S. Bates, and P. K. Quinn,
Physical properties of marine boundary layer aerosol particles
of the mid-Pacific in relation to sources and meteorological
transport, J. Geophys. Res., in press, 1995.
Draxler, R. R., Hybrid single-particle Lagrangian integrated
trajectories (HY-SPLIT): Version 3.0, User's Guide and Model
Description, Tech. Rep. ERL ARL-195, Natl. Oceanic and
Atmos. Admin., Silver Springs, Md., 1992.
Hegg, D. A., D. S. Covert, and V. N. Kapustin, Modeling a case of
particle nucleation in the marine boundary layer, J. Geophys.
Res., 97, 9851-9857, 1992.
Holland, H. D., The Chemistry of the Atmosphere and Oceans, p.
154, John Wiley, New York, 1978.
Hoppel, W. A., G. M. Frick, J. W. Fitzgerald, and R. E. Larson,
Marine boundary layer measurements of new particle formation
and the effect which nonprecipitating clouds have on the aerosol
size distribution, J. Geophys. Res., 99, 14,443-14,459, 1994.
Keady, P. B., F. R. Quant, and G. S. Sem, Differential mobility
particle sizer: A new instrument for high resolution aerosol size
distribution measurements below 1 pm, TSI Q., 9, 3-11, 1983.
Kent, G. S., et al., Modeling atmospheric aerosol backscatter at
CO 2 laser wavelengths, 1, Aerosol properties, modeling
techniques, and associated problems, Appl. Opt., 22, 1655-
1665, 1983.
Langner, J. and H. Rodhe, A global three-dimensional model of the
tropospheric sulfur cycle, J. Atmos. Chem., 13, 225-263, 1991.
Liu, B. Y. H., and K. W. Lee, Efficiency of membrane and
Nuclepore filters for submicrometer aerosols, Environ. Sci.
Technol., 10, 345-350, 1976.
Liu, B. Y. H., and D. Pui, On the performance of the electrical
aerosol analyzer, J. Aerosol Sci., 6, 249-264, 1975.
Marshall, S. F., Measurement-derived radiative transfer parameters
for the aerosol climate forcing problem, M.S. dissertation, Univ.
of Washington, Seattle, 1994.
Meszaros, A., and K. Vissy, Concentrations, size distribution and
chemical nature of atmospheric aerosol particles in remote
ocean areas, J. Aerosol Sci., 5, 101-109, 1974.
Mclnnes, L. M., D. S. Covert, and B. M. Baker, The number of
sea-salt sulfate and carbonaceous particles in the marine
atmosphere: Measurements consistent with the ambient size
distribution, in preparation, 1995a.
McInnes, L. M., P. K. Quinn, D. S. Covert, and T. L. Anderson,
Gravimetric analysis, ionic composition, and associated water
mass of the marine aerosol, Atmos. Environ., in press, 1995b.
Merrill, J., E. Arnold, M. Leinen, and C. Weaver, Mineralogy of
aeolian dust reaching the North Pacific Ocean, 2, Relationship
of mineral assemblages to atmospheric transport patterns, J.
Geophys. Res., 99, 21,025-21,032, 1994.
Monahan, E. C., The ocean as a source for atmospheric particles,
in The Role of Air-Sea Exchange in Geochemical Cycling,
edited by P. Buat-Menard, pp. 331-362, D. Reidel, Norwell,
Mass., 1986.
O'Dowd, C. D., and M. H. Smith, Physicochemical properties of
aerosols over the North Atlantic: Evidence for wind-speed-
related submicron sea-salt aerosol production, J. Geophys. Res.,
98, 1137-1149, 1993.
Plass-Dulmer, C., R. Koppmann, M. Ratte, and J. Rudolph, Light
nonmethane hydrocarbons in seawater, Global Biogeochem.
Cycles, 9, 79-100, 1995.
Pszenny, A. A. P., A. J. Castelie, R. A. Duce, and J. N. Galloway,
A study of the sulfur cycle in the Antarctic marine boundary
layer, J. Geophys. Res., 94, 9818-9830, 1989.
Quinn, P. K., T. S. Bates, J. E. Johnson, D. S. Covert, and R. J.
Charlson, Interactions between sulfur and reduced nitrogen
cycles over the central Pacific Ocean, J. Geophys. Res., 95,
16,405-16,416, 1990.
Quinn, P. K., D. S. Covert, T S. Bates, V. N. Kapustin, D.C.
Ramsey-Bell, and L. M. Mclnnes, Dimethylsulfide/cloud
condensation nuclei/climate system: Relevant size-resolved
measurements of the chemical and physical properties of
atmospheric aerosol particles, J. Geophys. Res., 98, 10,411-
10,427, 1993.
Quinn, P. K., S. F. Marshall, T S. Bates, D. S. Covert, and V. N.
Kapustin, Comparison of measured and calculated aerosol
properties relevant to the direct radiative forcing of tropospheric
sulfate aerosol on climate, J. Geophys. Res., 100, 8977-8992,
1995.
Rau, J. A., and M. A. K. Khalil, Anthropogenic contributions to the
carbonaceous content of aerosols over the Pacific Ocean, Atmos.
Environ., 27A, 1297-1307, 1993.
Reineking, A., and J. Porstendorfer, Measurements of particle loss
functions in a differential mobility analyzer for different flow
rates, Aerosol Sci. Technol., 5, 483-487, 1986.
Ruby, M. G., and A. P. Waggoner, Intercomparison of integrating

QUINN ET AL.: CHEMICAL AND OPTICAL PROPERTIES OF AEROSOLS 6951
nephelometer measurements, Environ. Sci. Technol., 15,
109-113, 1981.
Savoie, D. L., and J. M. Prospero, Particle size distribution of
nitrate and sulfate in the marine atmosphere, Geophys. Res.
Lett., 9, 1207-1210, 1982.
Sloane, C. S., J. Watson, J. Chow, L. Pritchett, and L. W. Richards,
Size-segregated fine particle measurements by chemical species
and their impact on visibility impairment in Denver, Atmos.
Environ., 25A, 1013-1024, 1991.
Stelson, A. W., Urban aerosol refractive index prediction by partial
molar refraction approach, Environ. Sci. Tech., 24, 1676-1679,
1990.
Stoiber, R. E., S. N. Williams, and B. Heubert, Annual contribution
of sulfur dioxide to the atmosphere by volcanos, J. Volcanol.
Geotherm. Res., 33, 1-8, 1987.
Tang, I. N., and H. R. Munkelwitz, Simultaneous determination of
refractive index and density of an evaporating aqueous solution
droplet, Aerosol. Sci. Technol., 15, 201-207, 1991.
Tang, I. N., and H. R. Munkelwitz, Water activities, densities, and
refractive indices of aqueous sulfate and nitrate droplets of
atmospheric importance, J. Geophys. Res., 99, 18,801-18,808,
1994.
ten Brink, H. M., S. E. Schwartz, and P. H. Daum, Efficient
scavenging of aerosol sulfate by liquid-water clouds, Atmos.
Environ., 21, 2035-2052, 1987.
Waggoner, A. P., A. J. Vanderpol, R. J. Charlson, S. Larsen, L.
Granat, and C. Tradgard, Sulfate light scattering ratio as an
index of the role of sulfur in the tropospheric optics, Nature,
261, 120-122, 1976.
Weiss, R. E., T. V. Larson, and A. P. Waggoner, In situ rapid-
response measurements of H2SO4/(NH4)2SO 4 aerosols in mral
Virginia, Environ. Sci. Technol., 16, 525-532, 1982.
Whitby, K. T., The physical characteristics of sulfur aerosols,
Atmos. Environ., 12, 135-159, 1978.
Whitby, K. T., and G. M. Sverdrup, California aerosols: Their
physical and chemical characteristics, Adv. Environ: Sci.
Technol., 10, 477-517, 1980.
Zang, Z., and B. Y. H. Liu, Performance of TS13760 condensation
nuclei counter at reduced pressures and flow rates, Aerosol Sci.
Technol., 15, 228-238, 1991.
Zhang, X., B. J. Turpin, P. H. McMurry, S. V. Hering, and M. R.
Stolzenburg, Mie theory evaluation of species contributions to
1990 wintertime visibility reduction in the Grand Canyon, J. Air
Waste Manage. Assoc., 44, 153-162, 1994.
T S. Bates, V. N. Kapustin, P. K. Quinn (corresponding author),
NOAA Pacific Marine Environmental Laboratory, 7600 Sand Point
Way N.E., Seattle, WA, 98115. (bates @pmel.noaa.gov; kapustin@
pmel.noaa.gov; [email protected])
D. S. Covert, Department of Atmospheric Sciences, University
of Washington, Seattle, WA 98195. ([email protected])
(Received June 22, 1995; revised October 27, 1995;
accepted November 1, 1995.)
