Chromatin modification and disease
Colin A Johnson
“Physicians consider that when they have discov-
ered the cause of disease, they have also discovered
the method of treating it.” Cicero, Tusculan Dis-
putations, III.x.23.
In the last few years, the exciting realisation
in the field of gene regulation is that transcrip-
tion factors can function by recruiting large,
multiprotein complexes which mediate several
types of chromatin modification and remodel-
ling events that alter the structure of chroma-
tin. Chromatin structure changes include post-
translational modifications of histones, DNA
methylation, remodelling of the chromatin, and
the maintenance of a heterochromatic or
euchromatic state. Most of these events are
brought about by enzymatic mechanisms. In
general, the catalytic subunits are only one
component of the complexes, with the distribu-
tion and localisation of the structural changes
dependent on targeting components. Many of
the catalytic components (sometimes called
coactivators and corepressors) interact with the
activator and repressor proteins that mediate
the actual process of transcriptional regulation.
Transcriptional dysregulation can therefore
arise from mutations that cause the loss or per-
turbation of chromatin modification or remod-
elling, which are now known to have an impor-
tant role in the pathogenesis of cancer and
other genetic diseases. Some of the proteins
that mediate these events are therefore novel
molecular targets for future treatments.
In eukaryotes, DNA is packaged by histone
proteins into nucleosomes, the fundamental
repeating structural unit of chromatin.
1
The
nucleosomal core particle consists of an
octomeric complex of core histones (two each
of H2A, H2B, H3, and H4) around which 147
bp of DNA is wrapped in 1.65 turns of a left
handed superhelix.
2
The minor and major
grooves of adjacent turns of the DNA superhe-
lix line up and form channels through which
the histone N-termini domains protrude from
the core. These regions are in the form of
“tails” that appear to lack secondary structure
3
and are subject to various enzyme catalysed,
post-translational modifications which aVect
their charge and can influence the degree of
chromatin compaction. The tightness with
which DNA is packaged into chromatin will
limit the binding and function of proteins that
mediate transcriptional regulation, and this will
therefore influence the transcriptional compe-
tence of any given gene in such a chromatin
environment.
24
Covalent post-translational acetylation and
deacetylation of specific lysine residues in the
histone N-termini is one of the most widely
studied chromatin modifications. In the past
four years there have been rapid advances in
identifying the enzymes and multiprotein com-
plexes that bring about histone acetylation (the
family of histone acetyltransferases or HAT
coactivators) and deacetylation (the histone
deacetylases or HDAC corepressors). This
review will focus on some of the clinical aspects
of this recent work on acetylation and the inti-
mate connection that it is now known to have
with the methylation of cytosine residues in
DNA. A third type of chromatin remodelling is
the direct physical repositioning or disruption
of nucleosomes mediated by a family of DNA
dependent ATPases. The connection between
this latter type of remodelling and either
histone acetylation or DNA methylation is
complicated, but progress is being made. For
example, the NuRD multiprotein complex (see
below, fig 1C) contains histone deacetylase and
chromatin remodelling activities, as well as the
methyl DNA binding protein MBD3, which
suggests that a profound interplay between
these modifications is required during gene
regulation. Therefore, it is probable that a par-
ticular pathogenesis may be caused by defects
in more than one type of chromatin modifica-
tion. Relevant pathologies and syndromes are
discussed in following sections and are summa-
rised in table 1.
Histone acetylation, protein acetylation,
and gene regulation
HISTONE DEACETYLASES AND COREPRESSOR
COMPLEXES
Deacetylation of histones is, in general, associ-
ated with repression of gene transcription, pre-
sumably because the highly positively charged
N-terminal tails of the core histones can now
interact with DNA on the nucleosome surface
and in the linker DNA.
56
In addition, the posi-
tively charged lysines in the H4 tail may inter-
act with the negative face of an H2A-H2B
dimer from a neighbouring nucleosome,
7
and
hence bring about further compaction of the
chromatin. Deacetylation is brought about by
the action of the histone deacetylases
(HDACs), which would therefore enhance
histone-histone interactions by maintaining the
positively charged (unmodified) state of lysines
in the histone tails. HDACs are now known to
be corepressor components of many multipro-
tein complexes that modify and remodel chro-
matin.
Targeting of complexes containing HDAC1
and HDAC2 is achieved by the interaction of
the repressor proteins Sin3A, Sin3B,
8
and
other Sin3 associated proteins (SAPs)
9
in a
large multiprotein complex that comprises at
least seven subunits
10
(fig 1A). The mammalian
Sin3 complex mediates repression for an
extensive and ever growing list of transcrip-
tional regulator proteins,
81011
which include
DNA binding components such as the Mad/
J Med Genet 2000;37:905–915 905
Chromatin and Gene
Expression Group,
Department of
Anatomy, University of
Birmingham,
Birmingham B15 2TT,
UK
Correspondence to:
Dr Johnson,
www.jmedgenet.com
Max heterodimer and nuclear hormone recep-
tors (see below, fig 1B). The members of the
Mad/Mxi1 family
12
are able to replace Myc in
the Myc/Max heterodimer, and can therefore
repress transcription at promoters with Myc
consensus DNA binding sites. Mutations in the
Sin3 interaction domains (SIDs) of Mad/Mxi1
can abolish binding of the corepressors to the
Sin3 proteins and hence HDACs, and this cor-
relates with the abolition of transcriptional
repression and anti-oncogenic activity.
13
In
addition, transfection studies have shown that
HDACs and Mad cooperate to repress cell
proliferation.
14
In a similar mechanism, the
transcriptional corepressor proteins N-CoR
(nuclear hormone receptor corepressor) and
SMRT (silencing mediator of retinoid and thy-
roid hormone receptor) target deacetylase
activity to non-liganded thyroid hormone and
retinoic acid nuclear receptors
15–17
and to
antagonist bound oestrogen and progesterone
receptors.
18
As discussed below, the presence of
receptor ligands, for example, retinoic acid,
appears to induce an exchange of the corepres-
sor complexes containing HDACs for those
with coactivator functions that contain histone
acetyltransferase (HAT) activities
18 19
(compare
figs 1B and 2A).
Histone deacetylases are also recruited by
the retinoblastoma protein pRb
20
(fig 1A), the
product of a tumour suppressor gene, and an
inhibitor of cell proliferation.
21
The inhibitory
action of pRb is the result, in part, of its ability
to bind to the E2F family of DNA binding
transcription factors, which results in the
sequestration of E2F and repression of E2F
target genes during the G1 phase of the cell
cycle.
22
The interaction is mediated by the A/B
pocket domain in pRb, and it is no coincidence
that the great majority of Rb mutations in
human tumours are located in this domain.
The pocket domain can also interact with a
variety of other cellular proteins, including viral
transforming oncoproteins (such as E1A from
adenovirus and SV40 large T
23
) and histone
deacetylases (HDAC1 and HDAC2) that share
the common LXCXE motif which allows
interactions with pocket proteins. However, the
interaction between Rb and either HDACs or
viral oncoproteins appears to be competitive,
and the Rb-HDAC1 interaction may be one of
the intracellular targets for these transforming
proteins. The Rb-HDAC interaction has been
analysed by transient transfection experiments,
which show that Rb and the HDACs cooperate
in repressing an E2F1-driven promoter,
24–26
and the repression exerted by Rb and other
pocket proteins during the G1 phase of the cell
cycle
27
can be reversed by treatment with
chemical inhibitors of HDACs (see below).
The HDAC inhibitors can also upregulate
some of the E2F target genes.
24
It is probable
that aberrant targeting of deacetylase activity
and incorrect chromatin remodelling are one
step in the process of transformation and
implies that these processes have a fundamen-
tal role in the suppression of carcinogenesis. In
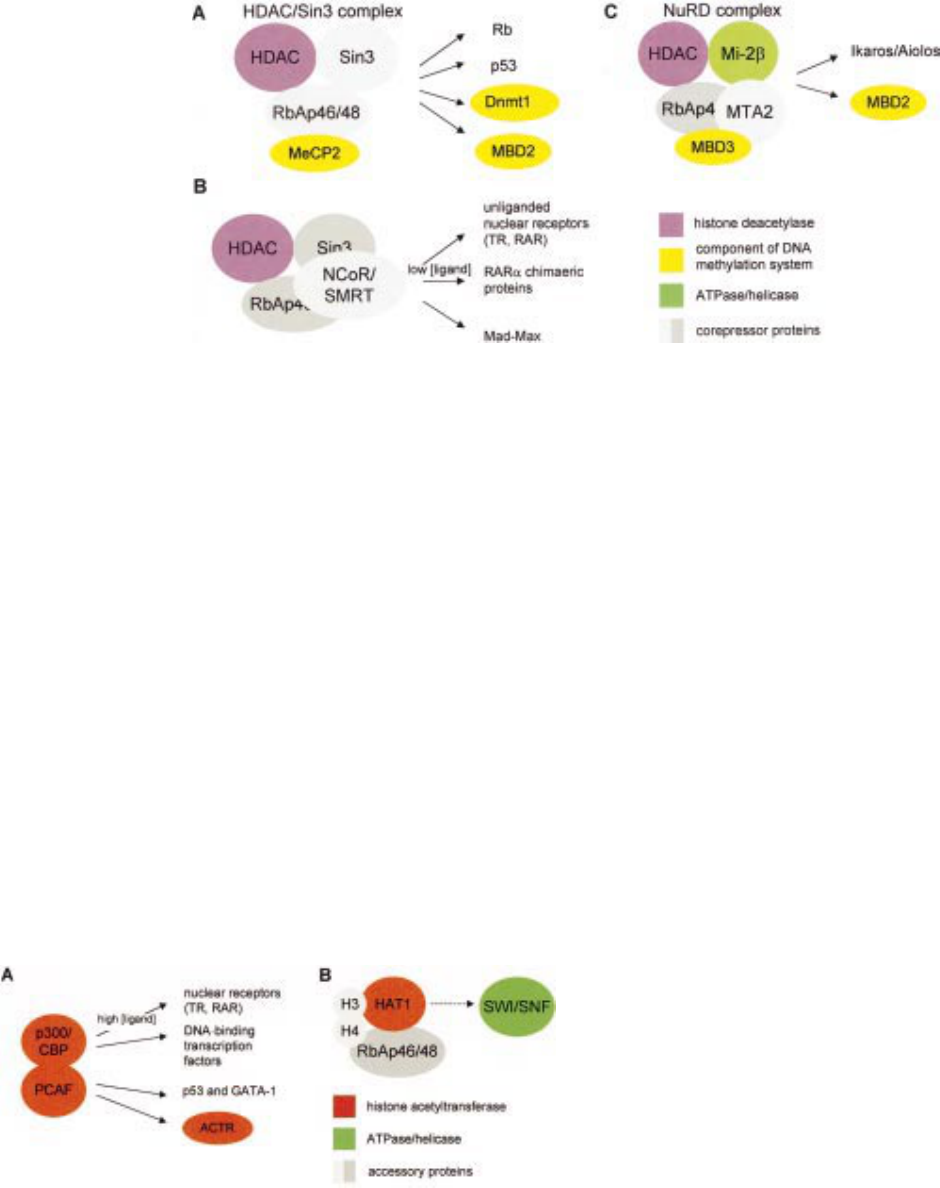
Figure 1 Schematic representation of multiprotein complexes that mediate chromatin modifications. Some of the known
components of the complexes are shown on the left, with arrows indicating additional interactions with other proteins on the
right (refer to main text for details). Histone deacetylases (HDAC) are shown in purple, components of the DNA
methylation system in yellow, and ATPase/helicase that mediates chromatin remodelling in green. Other corepressor
accessory proteins in the complexes (panels A-C) are shown in grey. (A) Components of the HDAC/Sin complex and
known interacting proteins. (B) Additional interactions of the HDAC/Sin3 complex, mediated by the corepressors
NcoR/SMRT, with unliganded nuclear receptors and leukaemogenic fusion proteins. Note that these interactions occur at
low concentrations, or in the absence of the receptor ligand. (C) Components and interactions of the NuRD complex.
Figure 2 Schematic representation of interactions mediated by histone acetyltransferases
(HATs). Unbroken arrows indicate known interactions of HATs with the proteins listed on
the right (refer to main text for details). Histone acetyltransferases are shown in red, the
SWI/SNF complex that mediates chromatin remodelling in green, and other accessory
proteins in grey. (A) Interactions of three human histone acetyltransferases. The p300/CBP
coactivator can interact with nuclear receptors at physiological concentrations of the receptor
ligand, with other transcription factors and with the histone acetyltransferase PCAF. PCAF
can also interact with the coactivator ACTR and transcription factors. (B) Putative
interactions of the human and/or yeast Hat1 protein with both RbAp48 and, through the
bromodomain, with histones H3 and H4. Histone H4 can also interact directly with both
RbAp46 and RbAp48. The bromodomain may also mediate an interaction with the yeast
SWI/SNF complex (broken arrow).
906 Johnson
www.jmedgenet.com

the case of pRb, histone modification is impli-
cated in the induction of cell cycle arrest which
may explain why the Rb gene is mutated in
almost all cancer cells.
In addition to E2F and HDACs, Rb has also
been shown to interact with other proteins that
regulate chromatin modifications: TAFII 250
is a transcription factor that has intrinsic
histone acetyltransferase (HAT) activity
28
;
BRG1 is a transcriptional activator and
ATPase/helicase that is a component of the
mammalian SWI/SNF chromatin remodelling
complex
29
; and Rb associated protein 48
(RbAp48) is a component of the chromatin
assembly factor CAF-1,
30
interacts with
HDAC1,
31
and is a component of the human
Hat1 acetyltransferase
32
(fig 2B). RbAp48, and
the related protein RbAp46, can bind directly
to an alpha helix in histone H4,
32
so it is likely
that these proteins mediate core histone
binding for the Sin3-HDAC complex, CAF-1
and Hat1.
A similar network of interactions that
regulate cell proliferation is also seen with the
HATs (see below) and for histone deacetylase
complexes other than the Sin3-HDAC com-
plex. For example, a novel multiprotein com-
plex has been isolated recently that contains
both nucleosome remodelling and histone
deacetylase activities (hence the NuRD com-
plex,
33
fig 1C). The NuRD complex contains,
in addition to HDAC1 and HDAC2, the
dermatomyositis specific autoantigen Mi-2â
34
that contains an ATPase/helicase domain of the
SWI/SNF type (see below). In addition, the
NuRD complex contains a protein, MTA2,
35
that is homologous to the metastasis associated
protein MTA1,
36
which is expressed at high
levels in several human cancer cell lines and
tissues. The NuRD complex can also be
recruited to DNA by specific DNA binding
factors, in a similar mechanism to the targeting
of the Sin3-HDAC complex. For example, two
determinants of the lymphoid lineage in T
cells, Ikaros and Aiolos, have been shown to
associate with the NuRD complex.
37
However,
Ikaros and Aiolos can also function as tran-
scriptional repressors during lymphocyte de-
velopment by recruiting the Sin3-HDAC com-
plex.
38
HISTONE DEACETYLASES AND LEUKAEMOGENIC
FUSION PROTEINS
The interaction of HDACs with chimeric
mutant proteins of the retinoic acid receptor is
one of the mechanisms that underlies the
molecular pathogenesis of acute promyelocytic
leukaemia (APL),
39 40
one of the best character-
ised forms of acute myeloid leukaemia
(AML).
41
In this disease, chromosomal translo-
cations create fusion proteins of retinoic acid
receptor-á and either PML (for promyelocytic
leukaemia) or, in rare cases, PLZF (for promye-
locytic leukaemia zinc finger).
42 43
In the case of
PML/RAR-á forms of APL, the fusion protein
retains the ability of the wild type nuclear
receptor to recruit the N-CoR/HDAC complex
(fig 1B) and to block haematopoietic diVeren-
tiation. However, physiological concentrations
of retinoic acid dissociate the corepressor com-
plex from wild type RAR-á (see above and fig
2A), so that it can therefore function as a tran-
scriptional activator. In contrast, the fusion
protein retains the ability to bind to the
corepressor complex under these conditions,
thereby constitutively repressing RAR-á target
genes. Treatment with higher, pharmacological
concentrations of the hormone overcomes this
interaction and converts the PML/RAR-á
fusion protein back into an activator. As a con-
sequence, cell proliferation is inhibited and
neutrophilic diVerentiation of neoplastic cells
is induced, which is the basis of diVerentiation
therapy of APL.
42
One of the target genes of
PML/RAR-á during RA induced diVerentia-
tion of APL cells is that encoding the
cyclin-CDK inhibitor p21
44
(also known as
WAF1 and CIP1), which exerts a G1 cell cycle
arrest in response to a variety of stimuli.
45
In contrast, cells expressing the PLZF/
RAR-á fusion are not sensitive to RA induced
diVerentiation, and patients with this type of
Table 1 Summary of human diseases in which a defect in chromatin modification and remodelling is believed to contribute to a clinical pathology. The
defects can arise from mutations in gene products, or by the aberrant recruitment of other proteins that are components of multiprotein complexes. Refer to
the main text for details
Type of chromatin
modification
Enzymes and other proteins that
mediate the chromatin modification
Mutated genes
implicated as a
cause of a
pathogenesis
Relevant proteins that
interact with the
enzymes etc
Clinical conditions and pathologies that
correlate with a listed mutation or
protein-protein interaction References
Histone acetylation Histone deacetylases
HDAC1/2 Rb pRb Neoplasia 24, 25, 26
p53 p53 Neoplasia 92
— Mi-2â Dermatomyositis and neoplasia 33, 34, 122
— ÌÔÁ2 Metastasis 35
RARá Fusion proteins Acute promyelocytic leukaemia 39, 40, 47, 48
Histone acetyltransferases
p300/CBP — E1A Transformation 82
MLL and MOZ Fusion proteins Acute myeloid leukaemia 94, 95
CBP — Rubinstein-Taybi syndrome 96
TAF II 250 — pRb Loss of tumour suppression? 28
Histone phosphorylation Rsk-2 kinase RSK2 Not known CoYn-Lowry syndrome 103
DNA methylation DNA methyltransferase DNMT3B Not known ICF syndrome 113, 114
MethylCpG binding proteins MECP2 HDAC/Sin3 Rett syndrome 125
MBD4 Not known Microsatellite unstable colon cancers 124
Chromatin remodelling ATPase/helicases
Mi-2â — NuRD complex Dermatomyositis and neoplasia 130, 131
ATRX ATRX Not known ATRX syndrome 134
BRG-1/hBRM — pRb Neoplasia 28
hSWI5/INI1 subunit of
SWI2/SNF2 complex
hSWI5/INI1 SWI2/SNF2 complex Malignant rhabdoid tumours,
chronic myeloid leukaemias
141, 142
Chromatin modification and disease 907
www.jmedgenet.com
APL respond poorly, if at all, to pharmacologi-
cal doses of the hormone.
46
The interaction
between PLZF/RAR-á and the corepressor
complex is resistant to retinoic acid because the
wild type PLZF protein can itself interact
directly with corepressors such as SMRT and
N-CoR.
39 40 47
This interaction is mediated by
the broad complex/tramtrack/bric a brac/
poxviruses and zinc finger (BTB/POZ) repres-
sion domain from PLZF,
48
which also allows
SMRT to interact with another BTB/POZ
oncoprotein, LAZ3/BCL6.
49
In contrast, other
transcriptional repressors that also contain the
BTB/POZ domain, such as the product of the
putative tumour suppressor gene HIC-1 (for
hypermethylated in cancer), do not recruit the
SMRT/N-CoR-histone deacetylase complex as
a general mechanism to repress transcription.
50
However, since HDAC inhibitors restore the
retinoid responses of RA resistant APL cell
lines,
39
clinical and cytogenetic remission of a
PLZF/RAR-á type of APL has been achieved
with a combination therapy of retinoic acid and
phenylbutyrate, an HDAC inhibitor.
51
Such
combination therapies may therefore be appli-
cable to other types of neoplastic diseases that
are associated with oncogenic repression of
gene transcription by histone deacetylases.
HISTONE DEACETYLASE INHIBITORS AND CANCER
CHEMOTHERAPY
Inhibitors of HDACs have also received
considerable attention as possible therapeutic
agents to induce growth arrest and terminal
diVerentiation in malignant cells and therefore
prevent the progression of cancers. HDAC
inhibitors can be classified on the basis of
structure and mode of inhibition. Reversible
inhibitors include n-butyric acid and other
related short chain fatty acids,
52 53
the microbial
antibiotic trichostatin A (TSA
54
), and hybrid
polar compounds such as suberoylanilide
hydroxamic acid (SAHA
55
). The carboxylic
and hydroxamic acid groups in this set of com-
pounds are likely to be specific ligands of a
catalytic zinc ion at the active site of HDAC
55 56
because a similar zinc binding site exists in a
prokaryotic homologue of the enzyme.
57
Irre-
versible inhibitors include trapoxin
58 59
and
trapoxin related natural products, such as
chlamydocin and HC toxin.
60
These com-
pounds are tetrapartite cyclic peptides with
2-amino-9,10-epoxy-8-oxodecanoic acid
(Aeo) as one residue. Aeo contains an epoxyke-
tone group that is isosteric with N-acetyl lysine
and is presumed to inhibit HDACs by binding
covalently and irreversibly to nucleophilic
groups in the active site of the enzymes. TSA,
trapoxin, and depudicin (a fungal metabolite
that resembles Aeo
61
) have all been shown to
revert the morphologies of oncogene trans-
formed cells and cells derived from tumours to
those with a normal cytoskeletal architec-
ture.
58 61 62
TSA and SAHA are also potent
inducers of transformed cell diVerentiation and
apoptosis.
55 63
A novel antibiotic and HDAC
inhibitor, FR901228,
64
can strongly inhibit
proliferation of tumour cells in vitro by arrest-
ing cell cycle transition at G1 and G2/M
phases, a property that is common between the
unrelated types of HDAC inhibitors.
65
For this
reason, HDAC inhibitors are being assessed as
therapeutic agents for cancer chemotherapy
66
and as adjuncts to established agents such as
retinoic acid (see above). In view of this, it is
interesting to note that n-butyric acid causes
growth inhibition, diVerentiation, and apopto-
sis in colon cancer derived cell lines.
67
Butyrate
is a natural fermentation product of certain
dietary fibres by anaerobic bacteria in the
lumen of the colon. The production of this
short chain fatty acid may be the key factor that
allows dietary fibre, for example fibre from
wheat, to protect against colon carcinogenesis.
The molecular mechanism that underlies this
protection is presumed to be inhibition of his-
tone deacetylase activity and induction of
histone hyperacetylation by butyrate.
68
Archer
et al
68
showed that butyrate induces expression
of the G1 cell cycle inhibitor p21 gene (see
above), presumably in response to hyper-
acetylation of the p21 promoter, which induces
growth arrest in colonic cancer cells. Trapoxin
has also been shown to increase p21 expression
in human tumour cells, with an increase of his-
tone H3 acetylation at the p21 promoter and
the induction of apoptosis in a cell line that
contained wild type p53.
69
HISTONE ACETYLTRANSFERASES AND
COACTIVATOR COMPLEXES
In parallel with the developments in our
understanding of HDACs, there have also been
the recent identification and functional analysis
of several human histone acetyltransferases
(HATs
70
) and the mechanisms by which
histone acetylation can activate transcription.
Histone acetylation, at lysines within the
N-terminal tails of H3 and H4, appears to be a
prerequisite for the process of transcriptional
activation in vivo.
71
The acetylation appears to
mediate chromatin remodelling (which makes
the chromatin more accessible to transcription
factors) by the specific, targeted interaction of
coactivator proteins that contain a bromodo-
main with acetyl lysines.
72
In particular, the
bromodomain of a yeast nuclear HAT is able to
bind in vitro to acetylated peptides of the H3
and H4 N-terminal tails
73
and the bromodo-
main is required for the subsequent recruit-
ment and coordination of in vivo remodelling
activity by the yeast SWI/SNF complex
74
(fig
2B).
Recent work has shown the intimate rela-
tionship between histone modifications and the
processes of cell proliferation, cell diVerentia-
tion, and oncogenesis. As discussed in previous
sections, nuclear receptors have a key role in
determining the balance of cell proliferation
and cell diVerentiation in response to extracel-
lular signals, such as the hormone retinoic acid.
In the absence of ligand, the receptors remain
constitutively bound to target promoters and
recruit a transcriptional repressor complex (see
above), which maintains the cell in a proliferat-
ing state. However, once the nuclear receptors
bind the ligand, the repressor complex is
replaced by an activator complex that induces
cell diVerentiation (fig 2A). Components of the
activator complexes include CREB binding
908 Johnson
www.jmedgenet.com
protein (CBP)
75 76
and the highly homologous
protein p300,
77 78
both of which are now known
to be histone acetyltransferases (HATs).
79 80
In
addition to CREB and nuclear hormone
receptors, the p300/CBP protein can also
interact with other DNA binding transcription
factors, such as c-Jun, c-Myb, c-Fos, and
MyoD
81
(fig 2A), as well as p300/CBP
associated factor (PCAF) that is itself a histone
acetyltransferase.
82
The studies by Yang et al
82
also show that the E1A oncoprotein stimulates
proliferation by disrupting the interaction of
p300/CBP and PCAF, which would normally
suppress cell growth. PCAF is also implicated
in nuclear receptor coactivation.
83 84
Two addi-
tional nuclear hormone receptor coactivators,
SRC-1 and ACTR, which are members of the
p160 family, are also HATs
85 86
(see below).
The human transcription factor TAF II 250, a
component of the TFIID complex that recog-
nises the TATA element at promoters, also
contains HAT activity.
28
A further level of complexity in the regula-
tion of gene expression by HATs is that the
enzymes can acetylate lysine groups of non-
histone proteins. For example, p300/CBP and
PCAF are able to acetylate transcription
factors such as p53 and components of the
general transcription machinery such as the
TFIIEâ subunit.
87 88
Acetylation of both p53
and the haematopoietic transcription factor
GATA-1 increased their DNA binding activi-
ties.
87 89
In the latter study, the acetylation of
GATA-1 in vivo was implicated in the activa-
tion of target genes. A similar mechanism of
regulation has been shown for the nuclear hor-
mone receptor coactivator and acetyltrans-
ferase ACTR, which can itself be acetylated by
p300/CBP.
90
As expected, hormone treatment
of cells caused an increase in histone acetyla-
tion at receptor target genes. However, this
eVect was transient because the subsequent
acetylation of ACTR led to dissociation of the
receptor-coactivator complex and down regu-
lation of transcription, and suggests that
non-histone protein acetylation is the key
element that autoregulates hormone induction.
In a separate study, the acetylation of E2F1 by
both PCAF and p300/CBP appears to enhance
the function of this transcription factor,
91
which would presumably stimulate the tran-
scription of target genes during S phase of the
cell cycle. As discussed above, the E2F
transcription factors repress transcription of
target genes during G1 phase by interacting
with the Rb tumour suppressor gene product
pRb and histone deacetylases (HDACs). In
addition, the p53 tumour suppressor gene
product has also been shown to repress
transcription during apoptosis by recruiting
the Sin3-HDAC complex,
92
and itself to
become acetylated at defined lysine residues in
vivo.
93
These observations suggest that tran-
scriptional regulation involves the addition and
removal of acetyl groups not only from histones
but also from other nuclear proteins, which has
important implications for the understanding
of cell growth and oncogenesis.
HISTONE ACETYLTRANSFERASE AND
LEUKAEMOGENIC FUSION PROTEINS
Transcriptional dysregulation by fusion pro-
teins that contain histone acetyltransferases
(HATs) has been implicated in leukaemogen-
esis
94 95
and in Rubinstein-Taybi syndrome
(RTS). RTS is a developmental disorder in
which patients have an increased incidence of
malignancies.
96
Microdeletions, translocations,
inversions, and point mutations in the CREB
binding protein (CBP) gene have been identi-
fied in patients with RTS.
96
CBP translocations
that correlate with acute myeloid leukaemia
(AML) include fusions to the MLL (also
known as ALL-1, see below) gene
94
and to the
MOZ gene.
95
MOZ is itself a histone acetyl-
transferase
95
and a chromosomal translocation
in AML can also fuse it to the nuclear receptor
coactivator TIF2.
97
There are, as yet, no known
chemical inhibitors of HATs that could there-
fore be assessed for AML therapy, in contrast
to the retinoic acid and HDAC inhibitor treat-
ments discussed above. It is likely that a nuclear
HAT inhibitor will be isosteric for the acetate
group, which would allow it to inactivate either
the catalytic site or the bromodomain of the
enzyme.
OTHER HISTONE MODIFICATIONS
The N-terminal tails of the core histones are
subject to other post-translational modifica-
tions, which include phosphorylation, methyla-
tion, ADP ribosylation, and ubiquitination.
98
Phosphorylation of histone H3 at serine 10 has
been the most extensively studied modification
to date. It occurs during mitosis in many cells
99
as a transient and rapid response after mitogen
stimulation, which correlates with the expres-
sion of c-fos and c-jun.
100
H3 phosphorylation
in response to epidermal growth factor (EGF)
appears to be mediated by the Rsk-2 kinase,
101
which is a member of the pp90
rsk
(ribosomal S6
kinase) family that is implicated in cell
proliferation and diVerentiation.
102
It is inter-
esting to note that mutations in Rsk-2 (but not
in any other kinase in this family) are associated
with CoYn-Lowry syndrome,
103
which is char-
acterised by severe psychomotor retardation,
facial and digital dysmorphism, and skeletal
deformations. However, it remains unclear
what role H3 phosphorylation has during
cellular response to mitogens and if it has a
direct involvement in gene regulation.
Methylation of histone H3 has been linked to
gene activation by the p160 family of coactiva-
tors, which include the histone acetyltrans-
ferases SRC-1 and ACTR (see above). Both of
these proteins, and a third member of this fam-
ily called GRIP1, have been shown to interact
with the coactivator associated arginine (R)
methyltransferase (CARM1), which has exten-
sive homology to other arginine methyltrans-
ferases.
104
CARM1 can also methylate histone
H3 in vitro, and enhances the expression of a
reporter gene in a transient transfection
experiment. Methylation of histones, or other
proteins in the transcription initiation com-
plex, may therefore be a gene regulatory mech-
anism that cooperates with histone and protein
acetylation.
Chromatin modification and disease 909
www.jmedgenet.com
DNA methylation and gene silencing
At the level of DNA modifications, methylation
of the C5 atom of cytosine residues is a power-
ful and prevalent mechanism for the repression
and inactivation of genes. Methylation of
promoter regions, on opposite DNA strands of
the dinucleotide sequence CpG, correlate, in
general, with transcriptional inhibition.
105
Hy-
drolytic deamination of 5'-methylcytosine to
thymine residues generates T-G mispairs,
which contribute to many germline point
mutations associated with human genetic
disease and other somatic mutations that lead
to cancer.
106 107
For this reason, CpG dinucle-
otides tend not to be found in coding regions,
but are clustered near the promoters of widely
expressed housekeeping genes, but remain
unmethylated at all levels of expression of the
gene. CpG islands can become de novo meth-
ylated during normal development (to silence
imprinted genes and the genes on the inactive
X chromosome of female mammals), but the
CpG islands of autosomal genes can also
become methylated as a result of in vitro cell
culture or neoplasia. It is possible that the
silencing of certain tumour suppressor genes
may, in part, be the consequence of de novo
methylation of an adjacent CpG island. For
example, the frequency with which promoter
methylation contributes to the gene inactiva-
tion is 33% for VHL and 84% for MLH1,in
von Hippel-Lindau (VHL) disease and micro-
satellite unstable colorectal tumours, respec-
tively.
108 109
A causal link between promoter
methylation and carcinogenesis is implied from
studies of transgenic mice with targeted
deletions of the DNA methyltransferase 1
(Dnmt1) gene, in which the formation of intes-
tinal polyps was suppressed.
110
Dnmt1 can also
recruit histone deacetylase activity and can
interact with HDAC1 in vitro.
111
Dnmt1 meth-
ylates DNA containing hemimethylated CpG
dinucleotides more eYciently than unmethyl-
ated DNA, and is therefore presumed to be the
major maintenance methyltransferase in
vivo.
112
Since Dnmt1 protein colocalises with
replication foci,
112
it will be interesting to see if
HDACs have a particular role at the replication
fork. Other mammalian DNA methyltrans-
ferases (Dnmt3a and b
113
) are likely to de novo
methylate promoters, with Dnmt3b specifically
required to methylate centromeric minor satel-
lite (in mouse embryonic stem cells).
114
Muta-
tions in one of the conserved catalytic domains
of DNMT3B, which presumably cause a partial
loss of function of the enzyme, are associated
with ICF syndrome (for immunodeficiency,
centromeric instability, facial anomalies).
114 115
ICF syndrome is characterised by immuno-
logical defects, hypomethylation, and instabil-
ity of centromeric heterochromatin, and facial
anomalies such as hypertelorism, epicanthic
folds, and macroglossia.
The mechanism by which methylated pro-
moters are inactivated has also become clearer
and appears to involve a long term remodelling
of the chromatin at the promoter. DNA meth-
ylation is able to reduce the binding aYnity of
sequence specific transcription factors or
recruit sequence specific DNA binding pro-
teins, such as methylated DNA binding protein
(MDBP),
116 117
which may act as transcriptional
repressors. However, a more general process
recruits sequence non-specific methyl-CpG
binding proteins (MeCPs) that exclude tran-
scription factors from the methylated pro-
moter.
10 118
It is now clear that the key molecu-
lar mechanism that underlies this repression
involves the recruitment of histone deacety-
lases (HDACs, see above and fig 1A). The
MeCP2 protein contains a methyl binding
domain (MBD), which allows it to bind to a
single, symmetrically methylated CpG site and
a transcriptional repression domain (TRD)
which recruits the Sin3-HDAC corepressor
complex.
119
This mechanism is now known to
be implicated in repression by members of the
MBD protein family, which were identified on
the basis of homology with the MBD domain
of MeCP2.
120
MBD2 forms the so called
MeCP1 complex, together with HDAC1,
HDAC2, and RbAp46 and 48,
10 121
and MBD3
is a component of the Mi2-NuRD deacetylase
complex
35 122
(see above). It is interesting to
note that MBD4 contains the canonical methyl
CpG binding domain in addition to a thymine
DNA glycosylase catalytic domain, which
binds preferentially to methyl CpG-TpG
mismatches in DNA.
123
It is therefore probable
that the function of the MBD4 enzyme is to
suppress mutation at methyl CpG, rather than
to act as a transcriptional repressor. Frameshift
mutations in MBD4 that would cause trunca-
tion of the protein between the MBD and gly-
cosylase domains, and hence cause a defect in
mismatch repair, correlate with over 40% of
microsatellite unstable sporadic colon can-
cers.
124
Mutations in the MECP2 gene, which
encodes the X linked methyl CpG binding
protein 2 (MeCP2, see above), are of particular
clinical significance. Genetic linkage analysis
has established that de novo missense muta-
tions in the methyl CpG binding domain
(MBD), and other mutations that disrupt the
transcriptional repression domain (TRD), are
a cause of Rett syndrome in about a quarter of
the sporadic patients studied.
125
Rett syndrome
is a progressive neurodevelopmental disorder
that occurs almost exclusively in females, and is
typified by the onset of autism, dementia,
ataxia, and loss of purposeful hand movements
from the ages of 6 to 18 months.
126
An
additional study has made the suggestion that
the eVect of MECP2 mutations may not be
limited to Rett syndrome, since heterozygote
females with skewed X inactivation patterns
may have a mild disease phenotype.
127
How-
ever, it is clear that the mechanism that under-
lies the pathogenesis of Rett implicates a
dysregulation in chromatin remodelling, al-
though it is not yet known if a similar
pathogenesis is seen for mutations in the MBD
family, or their partners in multiprotein
complexes.
Chromatin remodelling and
carcinogenesis
A further causal link between epigenetic
dysregulation and carcinogenesis is provided
910 Johnson
www.jmedgenet.com
by defects in ATP utilising chromatin remodel-
ling complexes. The role of these complexes is
to physically reposition or disrupt nucleosomes
by altering histone-DNA contacts.
128 129
The
ATPase/helicase subunits of the complexes are
members of three related families: homologues
of yeast SWI2/SNF2 (for mating type switch/
sucrose non-fermenting 2), the Mi-2 family
(also known as the CHD family), and the ISWI
(imitation SWI) family.
As discussed above, the dermatomyositis
specific autoantigen and ATPase/helicase
Mi-2â (also called CHD4) is a component of
the NuRD complex
33
that contains the histone
deacetylases HDAC1 and HDAC2 (fig 1C).
This observation suggests that chromatin
remodelling by the ATPase activity makes the
histone tails more accessible to the NuRD
deacetylases, with the subsequent formation of
a repressive chromatin structure. However,
neither the eVect of NuRD mediated histone
deacetylation on gene expression nor the
targets of this type of chromatin remodelling
are known, although the MBD3 component of
the complex may recruit it to extensive, meth-
ylated regions of the genome such as hetero-
chromatin. Since 15% to 30% of patients with
dermatomyositis develop cancer,
130 131
it is
therefore probable that changes in chromatin
modification and remodelling can aVect cell
proliferation.
It is interesting to note that the Mi-2â
protein contains two cysteine rich PHD (plant
homeodomain)/zinc finger regions and two
chromodomains, in addition to the helicase/
ATPase domain. Homeodomains and chromo-
domains are common features of other tran-
scriptional regulators that bind to
chromatin,
72 132
which include the DNMT3
family of DNA methyltransferases
112
and
ALL-1 (also known as MLL, HRX, or HTRX,
see below).
133
Another such protein is ATRX
(for á thalassaemia/retardation on the X chro-
mosome) which, like Mi2â, also contains
PHD-like fingers and other domains that clas-
sify it as a member of the ATPase/helicase
superfamily. The functional importance of the
PHD-like fingers is shown by the observation
that two thirds of all mutations that cause
ATRX syndrome lie in this region.
134
ATRX
syndrome comprises a severe form of mental
retardation, characterised by the presence of á
thalassaemia, urogenital abnormalities, and
facial dysmorphism.
135 136
In view of the interac-
tion of Mi2â protein with HDAC1,
33
it is prob-
able that PHD-like domains in other transcrip-
tional regulators also mediate the same
interaction. In addition, mutations in ATRX
are associated with changes in DNA methyla-
tion patterns at highly repeated sequences,
such that rDNA repeat arrays are hypomethyl-
ated whereas others (the Y specific repeat
DYZ2) are hypermethylated.
137
This observa-
tion implies a link between chromatin remodel-
ling, mediated by the ATRX protein, and the
DNA methylation system. However, it is
unclear if ATRX functions as a transcriptional
coactivator (similar to the SWI2/SNF2 pro-
teins) to increase the expression of a compo-
nent of the DNA methylation system, or if it
increases the accessibility of chromatin to a
DNA methyltransferase.
The human SWI/SNF complex has been
found to act as a coactivator for several nuclear
receptors
138
and appears to be both recruited to
specific regions of chromatin by DNA binding
proteins and to remodel promoter regions to
facilitate the binding of other factors.
129 139
The
two human homologues of SWI/SNF2, BRG-1
and hBRM, are also implicated in transcrip-
tional repression, since both can interact with
the tumour suppressor gene product Rb.
20 140
As discussed above, Rb regulates cell cycle
progression by inhibiting the activity of the
transcriptional activator E2F and recruiting
histone deacetylases. Transient transfection of
BRG-1 or hBRM into cell lines that do not
express these proteins but that do express Rb
leads to growth arrest
29
and, furthermore,
fibroblasts transformed with ras and lacking
endogenous expression of BRM revert to a
flattened, growth arrested phenotype after
reintroduction of hBRM.
140
These observations
establish the link between chromatin remodel-
ling by SWI/SNF and control of the cell cycle.
A further connection is provided by another
subunit of the SWI/SNF complex, called
hSNF5/INI1. Genetic linkage has identified
biallelic deletions or mutations in the hSWF5/
INI1 gene to be responsible for malignant
rhabdoid tumours, which are very aggressive
cancers of early childhood that tend to occur in
the kidney, brain, and soft tissues.
141
Another
study has shown that deletions of hSNF5/INI1
can be aquired during leukaemogenesis in
patients with chronic myeloid leukaemia.
142
Maintenance of chromatin states and
carcinogenesis
Recent work has shown that the maintenance
of active and inactive chromatin states is an
important determinant of gene expression. In
Drosophila, the coordinated expression of the
homeotic genes determines segmentation and
body plan along the anterior-posterior axis.
The maintenance of this expression through-
out development is mediated by the Polycomb-
group (Pc-G) repressor proteins and the
trithorax-group (trx-G) activator proteins.
143
Many of the polycomb- and trithorax-group
proteins contain the SET domain (for
Suvar3-9, Enhancer-of-zeste, Trithorax), that
is conserved in a number of mammalian
homologues. ALL-1, for example, is the human
homologue of Drosophila Trithorax protein,
which is a positive regulator of homeotic gene
expression. A similar role is implied for the
mammalian MLL/ALL-1 because hetero-
zygous mll
+/-
mice have defects in axial skeletal
development and haematopoiesis, which arise
from alterations in the pattern of Hox gene
expression.
144
The ALL-1 gene, at the 11q23
locus, is one of the most common targets of
chromosomal translocations in acute lym-
phocytic leukaemia and other acute leukae-
mias,
145
and there are at least 30 partner genes
that produce in frame leukogenic fusion
proteins with ALL-1/MLL, including the
histone acetyltransferase CBP
94
(see above). In
general, the C-terminal SET domain of
Chromatin modification and disease 911
www.jmedgenet.com
ALL-1/MLL is lost during chromosomal
translocation. It is interesting to note that the
hSNF5/INI1 component of the SWI/SNF
complex (see above) has been shown to interact
with the SET domain of ALL-1.
146
One of the
consequences of ALL-1/MLL translocations
would be the inability to recruit SWI/SNF, and
it is therefore possible that the transformation
of haematopoietic cells involves the dysregula-
tion of chromatin remodelling, as well as the
aberrant expression of MLL/ALL-1 target
genes.
Mammalian polycomb-group homologues
are thought to be negative regulators of home-
otic gene expression. EED (for embryonic
ectoderm development) is required at a very
early stage in embryonic development and is
the only known homologue of the Drosophila
extra sex combs (esc) Pc-G protein. Disruption
of the eed gene in mice causes defects in
anterior mesoderm production, followed by
death at day 8.5 of gestation.
147
In addition, the
histone deacetylases HDAC1 and HDAC2
have been shown to interact with EED, but not
with other vertebrate Pc-G proteins.
148
This
result again underlines the complex and subtle
interplay between the separate mechanisms of
gene regulation.
Conclusion
In the past few years, the exciting progress in
the field of gene regulation has made it clear
that chromatin is not just a static structure, but
that it has a pivotal role in regulating transcrip-
tion. Chromatin has a dynamic structure that
can be modulated during cell diVerentiation
and transformation, and the nucleosome is a
substrate that can both receive and transmit
intracellular signals. In addition, chromatin has
the capacity to encode epigenetic information
on levels of gene expression, which is inde-
pendent of the genetic information encoded by
the sequence of genes.
149
A parallel develop-
ment is the realisation that many transcription
factors can function by recruiting large, multi-
protein complexes that mediate several types of
chromatin modification and remodelling
events. It is therefore not surprising that muta-
tions that cause loss or perturbation of
chromatin modification and remodelling ac-
tivities will cause changes in gene expression
levels. This points to abnormal epigenetic
regulation as a general mechanism that under-
lies human carcinogenesis and the pathogen-
esis of other genetic diseases (see table 1 for a
summary). For example, the types of mutations
that are associated with most classes of acute
myeloid leukaemia are typified by balanced
chromosome translocations that result in the
expression of chimaeric proteins. Most of these
chimaeric proteins involve the in frame fusion
of transcriptional regulator that results in tran-
scriptional dysregulation. Some of the proteins
that mediate chromatin modification and
remodelling are therefore novel molecular
targets for future treatments of cancer and
other genetic diseases.
One of the most intriguing aspects of this
recent work is the probable interplay between
modification and remodelling events. Future
work will undoubtedly uncover further mecha-
nistic links between distinct gene regulation
systems. But a hint of this interplay is seen with
the possible interactions of histone deacetylase.
As discussed above, HDAC1 and HDAC2 can
interact with the DNA methyltransferase
Dnmt1, the polycomb-like protein EED, and
the ATPase/helicase Mi-2â. To add to this
complexity, it is not known if other histone and
protein post-translational modifications (nota-
bly histone phosphorylation and methylation)
can modulate the eVects of chromatin modifi-
cation and remodelling, or if they participate in
other gene regulation systems of their own.
Another important aspect is a description and
understanding of the signal transduction path-
ways that use chromatin as a target, and how
these signals can mediate changes in gene
expression. For example, a very recent develop-
ment has shown that recombinant yeast and
mouse Sir2 proteins, and other yeast Sir2
homologues, are novel, NAD dependent his-
tone deacetylases.
150 151
In yeast, Sir2 is a
mediator of transcriptional silencing at the het-
erochromatic regions associated with silent
mating loci, telomeres, and ribosomal DNA.
152
However, the absolute requirement of NAD for
the deacetylation reaction suggests that this
form of chromatin modification can be regu-
lated by the metabolic levels of NAD and
NADH, and hence by calorific intake.
153
The author would like to thank Doug Higgs, Institute of
Molecular Medicine, Oxford and members of the Chromatin
and Gene Expression Group for suggestions and critical evalu-
ation of the manuscript.
1 Kornberg RD, Lorch Y. Twenty-five years of the nucleo-
some, fundamental particle of the eukaryote chromosome.
Cell 1999;98:285-94.
2 Paranjape SM, Kamakaka R, Kadonaga J. Role of chromatin
structure in the regulation of transcription by RNA
polymerase II. Annu Rev Biochem 1994;63:265-97.
3 Luger K, Mader AW, Richmond RK, Sargent DF,
Richmond TJ. Crystal structure of the nucleosome core
particle at 2.8 A resolution. Nature 1997;389:251-60.
4 Workman JL, Kingston RE. Alteration of nucleosome struc-
ture as a mechanism of transcriptional regulation. Annu
Rev Biochem 1998;67:545-79.
5 Usachenko SI, Bavykin SG, Gavin IM, Bradbury EM. Rear-
rangement of the histone H2A C-terminal domain in the
nucleosome. Proc Natl Acad Sci USA 1994;91:6845-9.
6 Walker IO. DiVerential dissociation of histone tails from
core chromatin. Biochemistry 1984;23:5622-8.
7 Luger K, Richmond TJ. The histone tails of the nucleo-
some. Curr Opin Genet Dev 1998;8:140-6.
8 Johnson CA, Turner BM. Histone deacetylases: complex
transducers of nuclear signals. Semin Cell Develop Biol
1999;10:179-88.
9 Zhang Y, Iratni R, Erdjument-Bromage H, Tempst P, Rein-
berg D. Histone deacetylases and SAP18, a novel polypep-
tide, are components of a human Sin3 complex. Cell 1997;
89:357-64.
10 Ng HH, Bird A. Histone deacetylases: silencers for hire.
Trends Biochem Sci 2000;25:121-6.
11 Struhl K. Histone acetylation and transcriptional regulatory
mechanisms. Genes Dev 1998;12:599-606.
12 Henriksson M, Luscher B. Proteins of the Myc network:
essential regulators of cell growth and diVerentiation. Adv
Cancer Res 1996;68:109-82.
13 Schreiber-Agus N, Chin L, Chen K, Torres R, Rao G,
Guida P, Skoultchi AI, DePinho RA. An amino terminal
domain of Mxi1 mediates anti-Myc oncogenic activity and
interacts with a homolog of the yeast transcriptional
repressor SIN3. Cell 1995;80:767-76.
14 Sommer A. Cell growth inhibition by the Mad/Max
complex through recruitment of histone deacetylase activ-
ity. Curr Biol 1997;7:357-65.
15 Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M,
Laherty CD, Torchia J, Yang WM, Brard G, Ngo SD, Davie
JR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosenfeld
MG. A complex containing N-CoR, mSin3 and histone
deacetylase mediates transcriptional repression. Nature
1997;387:43-8.
16 Alland L, Muhle R, Hou HJ, Potes J, Chin L, Schreiber-
Agus N, DePinho R. Role for N-CoR and histone deacety-
lase in Sin3-mediated transcriptional repression. Nature
1997;387:49-55.
912 Johnson
www.jmedgenet.com
17 Nagy L, Kao H, Chakravarti D, Lin R, Hassig C, Ayer D,
Schreiber S, Evans R. Nuclear receptor repression
mediated by a complex containing SMRT, mSin3A and
histone deacetylase. Cell 1997;89:373-80.
18 Xu L, Glass CK, Rosenfeld MG. Coactivator and corepres-
sor complexes in nuclear receptor function. Curr Opin
Genet Dev 1999;9:140-7.
19 Grunstein M. Histone acetylation in chromatin structure
and transcription. Nature 1997;389:349-52.
20 Brehm A, Kouzarides T. Retinoblastoma protein meets
chromatin. Trends Biochem Sci 1999;24:142-5.
21 Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Wein-
berg RA. Regulation of retinoblastoma protein functions by
ectopic expression of human cyclins. Cell 1992;70:993-
1006
22 Weinberg RA. E2F and cell proliferation: a world turned
upside down. Cell 1996;85:457-9.
23 Kaelin WG, Ewen ME, Livingston DM. Definition of the
minimal simian virus 40 large T- and adenovirus E1A-
binding domain in the retinoblastoma gene product. Mol
Cell Biol 1990;10:3761-9.
24 Luo RX, Postigo AA, Dean DC. Rb interacts with histone
deacetylase to repress transcription. Cell 1998;92:463-73.
25 Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ,
Kouzarides T. Retinoblastoma protein recruits histone
deacetylase to repress transcription. Nature 1998;391:597-
601.
26 Magnaghi-Jaulin L, Groisman R, Naguibneva I, Robin P,
Lorain S, Le Villain JP, Troalen F, Trouche D, Harel-Bellan
A. Retinoblastoma protein represses transcription by
recruiting a histone deacetylase. Nature 1998;391:601-5.
27 Ferreira R, Magnaghi-Jaulin L, Robin P, Harel-Bellan A,
Trouche D. The three members of the pocket proteins
family share the ability to repress E2F activity through
recruitment of a histone deacetylase. Proc Natl Acad Sci
USA 1998;95:10493-8.
28 Mizzen CA, Yang XJ, Kokubo T, Brownell JE, Bannister AJ,
Owen-Hughes T, Workman J, Wang L, Berger SL,
Kouzarides T, Nakatani Y, Allis CD. The TAF(II)250
subunit of TFIID has histone acetyltransferase activity. Cell
1996;87:1261-70.
29 Dunaief JL, Strober BE, Guha S, Khavari PA, Alin K,
Luban J, Begemann M, Crabtree GR, GoV SP. The retino-
blastoma protein and BRG1 form a complex and cooperate
to induce cell cycle arrest. Cell 1994;79:119-30.
30 Roth SY, Allis CD. Histone acetylation and chromatin
assembly: a single escort, multiple dances? Cell 1996;87:5-
8.
31 Taunton J, Hassig CA, Schreiber SL. A mammalian histone
deacetylase related to the yeast transcriptional regulator
Rpd3p. Science 1996;272:408-11.
32 Verrault A, Kaufman PD, Kobayashi R, Stillman B. Nucleo-
somal DNA regulates the core-histone binding subunit of
the human Hat1 acetyltransferase. Curr Biol 1998;8:96-
108.
33 Zhang Y, LeRoy G, Seelig HP, Lane WS, Reinberg D. The
dermatomyositis-specific autoantigen Mi2 is a component
of a complex containing histone deacetylase and a nucleo-
some remodeling activities. Cell 1998;95:279-89.
34 Seelig H, Moosbrugger I, Ehrfeld H, Fink T, Renz M,
Genth E. The major Mi-2 autoantigen is a presumed heli-
case involved in transcriptional activation. Arthritis Rheum
1995;38:1389-99.
35 Zhang Y, Ng HH, Erdjument-Bromage H, Tempst P, Bird
A, Reinberg D. Analysis of the NuRD subunits reveals a
histone deacetylase core complex and a connection with
DNA methylation. Genes Dev 1999;13:1924-35.
36 Toh Y, Pencil SD, Nicolson GL. A novel candidate
metastasis-associated gene, mta1,diVerentially expressed in
highly metatastic mammary adenocarcinoma cell lines:
cDNA cloning, expression and protein analysis. J B iol
Chem 1994;269:22958-63.
37 Kim J, Sif S, Jones B, Jackson A, Koipally J, Heller E,
Winandy S, Viel A, Sawyer A, Ikeda T, Kingston R, Geor-
gopoulos K. Ikaros DNA-binding proteins direct formation
of chromatin remodeling complexes in lymphocytes.
Immunity 1999;10:345-55.
38 Koipally J, Renold A, Kim J, Georgopoulos K Repression by
Ikaros and Aiolos is mediated through histone deacetylase
complexes. EMBO J 1999;18:3090-100.
39 Lin RJ, Nagy L, Inoue S, Shao W, Miller WH Jr, Evans RM.
Role of the histone deacetylase complex in acute promyelo-
cytic leukaemia. Nature 1998;391:811-14.
40 Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti
V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I,
Seiser C, Grignani F, Lazar MA, Minucci S, Pelicci PG.
Fusion proteins of the retinoic acid receptor-á recruit
histone deacetylase in promyelocytic leukaemia. Nature
1998;391:815-18.
41 Redner RL, Wang J, Liu JM. Chromatin remodelling and
leukaemia: new therapeutic paradigms. Blood 1999;94:417-
28.
42 Grignani F, Fagioli M, Alcalay M, Longo L, Pandolfi PP,
Donti E, Biondi A, Lo Coco F, Grignani F, Pelicci PG.
Acute promyelocytic leukemia: from genetics to treatment.
Blood 1994;83:10-25.
43 Golub TR. The genetics of AML: an update. Blood
1999;(suppl):102-11.
44 Casini T, Pelicci PG. A function of p21 during promyelo-
cytic leukaemia cell diVerentiation independent of CDK
inhibition and cell cycle arrest. Oncogene 1999;18:3235-43.
45 Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks
T, Hannon GJ. Radiation-induced cell cycle arrest
compromised by p21 deficiency. Nature 1995;377:552-7.
46 Licht JD, Chomienne C, Goy A, Chen A, Scott AA, Head
DR, Michaux JL, Wu Y, DeBlasio A, Miller WH Jr,
Zelenetz AD, Willman CL, Chen Z, Chen S-J, Zelent A,
Macintyre E, Veil A, Cortes J, Kantarjian H, Waxman S.
Clinical and molecular characterisation of a rare syndrome
of acute promyelocytic leukaemia associated with translo-
cation (11;17) Blood 1995;85:1083-94.
47 Hong SH, David G, Wong CW, Dejean A, Privalsky ML.
SMRT corepressor interacts with PLZF and with the
PML-retinoic acid receptor alpha (RARalpha) and PLZF-
RARalpha oncoproteins associated with acute promyelo-
cytic leukaemia. Proc Natl Acad Sci USA 1997;94:9028-33.
48 Li JY, English MA, Ball HJ, Yeyati PL, Waxman S, Licht JD.
Sequence-specific DNA binding and transcriptional regu-
lation by the promyelocytic leukemia zinc finger protein. J
Biol Chem 1997;272:22447-55.
49 Dhordain P, Albagli O, Lin RJ, Ansieau S, Quief S, Leutz A,
Kerckaert JP, Evans RM, LePrince D. Corepressor SMRT
binds the BTB/POZ repressing domain of the LAZ3/BCL6
oncoprotein. Proc Natl Acad Sci USA 1997;94:10762-7.
50 Deltour S, Guerardel C, Leprince D. Recruitment of
SMRT/N-CoR-mSin3A-HDAC-repressing complexes is
not a general mechanism for BTB/POZ transcriptional
repressors: the case of HIC-1 and gammaFBP-B. Proc Natl
Acad Sci USA 1999;96:14831-6.
51 Warrell RP Jr, He LZ, Richon V, Calleja E, Pandolfi PP.
Therapeutic targeting of transcription in acute promyelo-
cytic leukemia by use of an inhibitor of histone deacetylase.
J Natl Cancer Inst 1998;90:1621-5.
52 Candido E, Reeves R, Davie JR. Sodium butyrate inhibits
histone deacetylation in cultured cells. Cell 1978;14:105-
13.
53 Sealy L, Chalkley R. The eVect of sodium butyrate on
histone modifications. Cell 1978;14:115-21.
54 Yoshida M, Hoshikawa Y, Koseki K, Mori K, Beppu T.
Structural specificity for biological activity of trichostatin
A, a specific inhibitor of mammalian cell cycle with potent
diVerentiation-inducing activity in Friend leukemia cells. J
Antibiot 1990;43:1101-6.
55 Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rif-
kind RA, Marks PA. A class of hybrid polar inducers of
transformed cell diVerentiation inhibits histone deacety-
lases. Proc Natl Acad Sci USA 1998;95:3003-7.
56 Hassig CA, Tong JK, Fleischer TC, Owa T, Grable PG,
Ayer DE, Schreiber SL. A role for histone deacetylase
activity in HDAC1-mediated transcriptional repression.
Proc Natl Acad Sci USA 1998;95:3519-24.
57 Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind
RA, Marks PA, Breslow R, Pavletich NP. Structures of a
histone deacetylase homologue bound to the TSA and
SAHA inhibitors. Nature 1999;401:188-93.
58 Itazaki H, Nagashima K, Sugita K, Yoshida H, Kawamura
Y, Yasuda Y, Matsumoto K, Ishii K, Uotani N, Nakai H.
Isolation and structural elucidation of new cyclotetrapep-
tides, trapoxins A and B, having detransformation activities
as antitumour agents. J Antibiot 1990;43:1524-32.
59 Kijima M, Yoshida M, Sugita K, Horinouchi S, Beppu T.
Trapoxin, an antitumour cyclic tetrapeptide, is an irrevers-
ible inhibitor of mammalian histone deacetylase. J Biol
Chem 1993;268:22429-35.
60 Brosch G, Ransom R, Lechner T, Walton JD, Loidl P. Inhi-
bition of maize histone deacetylases by HC toxin, the host-
specific toxin of Cochliobolus carbonum. Plant Cell 1995;7:
1941-50.
61 Kwon HJ, Owa T, Hassig CA, Shimada J, Schreiber SL.
Depudicin induces morphological reversion of transformed
fibroblasts via the inhibition of histone deacetylase. Proc
Natl Acad Sci USA 1998;95:3356-61.
62 Hoshikawa Y, Kwon HJ, Yoshida M, Horinouchi S, Beppu
T. Trichostatin A induces morphological changes and gel-
solin expression by inhibiting histone deacetylase in human
carcinoma cell lines. Exp Cell Res 1994;214:189-97.
63 Yoshida M, Nomura S, Beppu T. EVects of trichostatins on
diVerentiation of murine erythroleukemia cells. Cancer Res
1987;47:3688-91.
64 Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S.
FR901228, a potent antitumour antibiotic, is a novel
histone deacetylase inhibitor. Exp Cell Res 1998;241:126-
33.
65 Yoshida M, Horinouchi S, Beppu T. Trichostatin A and
trapoxin: novel chemical probes for the role of histone
acetylation in chromatin structure and function. BioEssays
1995;17:423-30.
66 Wang J, Saunthararajah Y, Redner RL, Liu JM. Inhibitors of
histone deacetylase releive ETO-mediated repression and
induce diVerentiation of AML1-ETO leukaemia cells.
Cancer Res 1999;59:2766-9.
67 Archer SY, Hodin RA. Histone acetylation and cancer. Curr
Opin Genet Develop 1999;9:171-4.
68 Archer SY, Meng S, Shei A, Hodin RA. p21/WAF1 is
required for butyrate-mediated growth inhibition of human
colon cancer cells. Proc Natl Acad Sci USA 1998;95:6791-
6.
69 Sambucetti LC, Fischer D, ZabludoV S, Kwon PO, Cham-
berlin H, Trogani N, Xu H, Cohen D. Histone deacetylase
inhibition selectively alters the activity and expression of
cell cycle proteins leading to specific chromatin acetylation
and antiproliferative eVects. J Biol Chem 1999;274:34940-
7.
70 Grant PA, Berger SL. Histone acetyltransferase complexes.
Semin Cell Develop Biol 1999;10:169-77.
71 Kuo MH, Zhou J, Jambeck P, Churchill ME, Allis CD. His-
tone acetyltransferase activity of yeast Gcn5p is required
Chromatin modification and disease 913
www.jmedgenet.com
for the activation of target genes in vivo. Genes Dev
1998;12:627-39.
72 Winston F, Allis CD. The bromodomain: a chromatin-
targeting module? Nat Struct Biol 1999;6:601-4.
73 Dhalluin C, Carlson JE, Zeng L, He C, Aggarwal AK, Zhou
MM. Structure and ligand of a histone acetyltransferase
bromodomain. Nature 1999;399:491-6.
74 Syntichaki P, Topalidou I, Thireos G. The Gcn5 bromodo-
main co-ordinates nucleosome remodelling. Nature 2000;
404:414-17.
75 Kamei Y, Xu L, Heinzel T, Torchia J, Kurokawa R, Gloss B,
Lin SC, Heyman RA, Rose DW, Glass CK, Rosenfeld MG.
A CBP integrator complex mediates transcriptional activa-
tion and AP-1 inhibition by nuclear receptors. Cell
1996;85:403-14.
76 Aarnisalo P, Palvimo JJ, Janne OA. CREB-binding protein
in androgen receptor-mediated signalling. Proc Natl Acad
Sci USA 1998;95:2122-7.
77 Chakravarti D, LaMorte VJ, Nelson MC, Nakajima T,
Schulman IG, Juguilon H, Montminy M, Evans RM. Role
of CBP/p300 in nuclear receptor signalling. Nature
1996;383:99-103.
78 Kraus WL, Kadonaga JT. p300 and oestrogen receptor
cooperatively activate transcription via diVerential en-
hancement of initiation and reinititation. Genes Dev
1998;12:331-42.
79 Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Naka-
tani Y. The transcriptional coactivators p300 and CBP are
histone acetyltransferases. Cell 1996;87:953-9.
80 Bannister AJ, Kouzarides T. The CBP co-activator is a his-
tone acetyltransferase. Nature 1996;384:641-3.
81 Janknecht R, Hunter T. Versatile molecular glue: transcrip-
tional control. Curr Biol 1996;6:951-4.
82 Yang XJ, Ogryzko VV, Nishikawa J, Howard BH, Nakatani
Y. A p300/CBP-associated factor that competes with the
adenoviral oncoprotein E1A. Nature 1996;382:319-24.
83 Blanco JC, Minucci S, Lu J, Yang XJ, Walker KK, Chen H,
Evans RM, Nakatani Y, Ozato K. The histone acetylase
PCAF is a nuclear receptor coactivator. Genes Dev
1998;12:1638-51.
84 Korzus E, Torchia J, Rose DW, Xu L, Kurokawa R, McIner-
ney EM, Mullen TM, Glass CK, Rosenfeld MG.
Transcription factor-specific requirements for coactivators
and their acetyltransferase functions. Science 1998;279:
703-7.
85 Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J,
Mizzen CA, McKenna NJ, Onate SA, Tsai SY, Tsai MJ,
O’Malley BW. Steroid receptor coactivator-1 is a histone
acetyltransferase. Nature 1997;389:194-8.
86 Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Nuclear
receptor coactivator ACTR is a novel histone acetyltrans-
ferase and forms a multimeric activation complex with
P/CAF and CBP/p300. Cell 1997;90:569-80.
87 Gu W, Roeder RG. Activation of p53 sequence-specific
DNA binding by acetylation of the p53 C-terminal
domain. Cell 1997;90:595-606.
88 Imhof A, Yang XJ, Ogryzko VV, Nakatani Y, WolVeAP,Ge
H. Acetylation of general transcription factors by histone
acetyltransferases. Curr Biol 1997;7:689-92.
89 Boyes J, Byfield P, Nakatani Y, Ogryzko V. Regulation of
activity of the transcription factor GATA-1 by acetylation.
Nature 1998;396:594-8.
90 Chen H, Lin RJ, Xie W, Wilpitz D, Evans RM. Regulation of
hormone-induced histone hyperacetylation and gene acti-
vation via acetylation of an acetylase. Cell 1999;98:675-86.
91 Martínez-Balbás MA, Bauer UM, Nielsen SJ, Brehm A,
Kouzarides T. Regulation of E2F1 activity by acetylation.
EMBO J 2000;19:662-71.
92 Murphy M, Ahn J, Walker KK, HoVman WH, Evans RM,
Levine AJ, George DL. Transcriptional repression by wild-
type p53 utilises histone deacetylases, mediated by interac-
tion with mSin3a. Genes Dev 1999;13:2490-501.
93 Liu L, Scolnick DM, Trievel RC, Zhang HB, Marmorstein
R, Halazonetis TD, Berger SL. p53 sites acetylated in vitro
by PCAF and p300 are acetylated in vivo in response to
DNA damage. Mol Cell Biol 1999;19:1202-9.
94 Sobulo OM, Borrow J, Tomek R, Reshmi S, Harden A, Sch-
legelberger B, Housman D, Doggett NA, Rowley JD,
Zeleznik-Le NJ. MLL is fused to CBP, a histone
acetyltransferase, in therapy-related acute myeloid leuke-
mia with a t(11;16)(q23;p13.3). Proc Natl Acad Sci USA
1997;94:8732-7.
95 Borrow J, Stanton VP Jr, Andresen JM, Becher R, Behm
FG, Chaganti RS, Civin CI, Disteche C, Dube I, Frischauf
AM, Horsman D, Mitelman F, Volinia S, Watmore AE,
Housman DE. The translocation t(8;16)(p11;p13) of acute
myeloid leukaemia fuses a putative acetyltransferase to the
CREB-binding protein. Nat Genet 1996;14:33-41.
96 Petrij F, Giles RH, Dauwerse HG, Saris JJ, Hennekam RC,
Masuno M, Tommerup N, van Ommen GJ, Goodman RH,
Peters DJ, Breuning MH. Rubinstein-Taybi syndrome
caused by mutations in the transcriptional co-activator
CBP. Nature 1995;376:348-51.
97 Carapeti M, Aguiar RC, Goldman JM, Cross NC. A novel
fusion between MOZ and the nuclear receptor coactivator
TIF2 in acute myeloid leukaemia. Blood 1998;91:3127-33.
98 Wu RS, Panusz HT, Hatch CL, Bonner WM. Histones and
their modifications. CRC Crit Rev Biochem 1986;20:201-
63.
99 Bradbury EM. Reversible histone modifications and the
chromosome cell cycle. BioEssays 1992;14:9-16.
100 Thomson S, Mahadevan L, Clayton AL. MAP kinase-
mediated signalling to nucleosomes and immediate-early
gene induction. Semin Cell Develop Biol 1999;10:205-14.
101 Sassone-Corsi P, Mizzen CA, Cheung P, Crosio C,
Monaco L, Jacquot S, Hanauer A, Allis CD. Requirement
of Rsk-2 for epidermal growth factor-activated phosphor-
ylation of histone H3. Science 1999;285:886-91.
102 Blenis J. Signal transduction via the MAP kinases: proceed
at your own RSK. Proc Natl Acad Sci USA 1993;90:5889-
92.
103 Trivier E, DeCesare D, Jacquot S, Pannetier S, Zackai E,
Young I, Mandel JL, Sassone-Corsi P, Hanauer A.
Mutations in the kinase Rsk-2 associated with CoYn-
Lowry syndrome. Nature 1996;384:567-70.
104 Chen D, Ma H, Hong H, Koh SS, Huang SM, Schurter
BT, Aswad DW, Stallcup MR Regulation of transcription
by a protein methyltransferase. Science 1999;284:2174-7.
105 Bird A. The essentials of DNA methylation. Cell 1992;70:
5-8.
106 Yang A, Jones P, Shibata A. The mutational burden of
5-methylcytosine. In: Russo V, Martienssen R, Riggs A,
eds. Epigenetic mechanisms of gene regulation. Cold Spring
Harbor, NY: Cold Spring Harbor Press, 1996:77-94.
107 Jones PA, Laird PW. Cancer epigenetics comes of age. Nat
Genet 1999;21:163-7.
108 Prowse AH, Webster AR, Richards FM, Richard S,
Olschwang S, Resche F, AVara NA, Maher ER. Somatic
inactivation of the VHL gene in Von Hippel-Lindau disease
tumours. Am J Hum Genet 1997;60:765-71.
109 Herman JG, Umar A, Polyak K, GraV JR, Ahuja N, Issa JP,
Markowitz S, Willson JK, Hamilton SR, Kinzler KW, Kane
MF, Kolodner RD, Vogelstein B, Kunkel TA, Baylin SB.
Incidence and functional consequences of hMLH1 pro-
moter hypermethylation in colorectal carcinoma. Proc Natl
Acad Sci USA 1998;95:6870-5.
110 Laird PW, Jackson-Grusby L, Fazeli A, Dickinson SL, Jung
WE, Li E, Weinberg RA, Jaenisch R. Suppression of intes-
tinal neoplasia by DNA hypomethylation. Cell 1995;81:
197-205.
111 Fuks F, Burgers WA, Brehm A, Hughes-Davies L,
Kouzarides T. DNA methyltransferase Dnmt1 associates
with histone deacetylase activity. Nat Genet 2000;24:88-91.
112 Bestor TH, Verdine GL. DNA methyltransferases. Curr
Opin Cell Biol 1994;6:380-9.
113 Okano M, Xie S, Li E. Cloning and characterisation of a
family of novel mammalian DNA (cytosine-5) methyltrans-
ferases. Nat Genet 1998;19:219-20.
114 Okano M, Bell DW, Haber DA and Li E. DNA
methyltransferases Dnmt3a and Dnmt3b are essential for
de novo methylation and mammalian development. Cell
1999;99:247-57.
115 Xu GL, Bestor TH, Bourc’his D, Hsieh CL, Tommerup
N, Bugge M, Hulten M, Qu X, Russo JJ, Viegas-Pequignot
E. Chromosome instability and immunodeficiency syn-
drome caused by mutations in a DNA methyltransferase
gene. Nature 1999;402:187-91.
116 Prendergast GC, ZiV EB. Methylation-sensitive sequence-
specific DNA binding by the c-Myc basic region. Science
1991;251:186-9.
117 Zhang XY, Ehrlich KC, Wang RY, Ehrlich M. EVect of
site-specific DNA methylation and mutagenesis on recog-
nition by methylated DNA-binding protein from human
placenta. Nucleic Acids Res 1985;14:8387-97.
118 Bird A, WolVe A. Methylation-induced repression—belts,
braces and chromatin. Cell 1999;99:451-4.
119 Nan X, Ng HH, Johnson CA, Laherty CA, Turner BM,
Eisenman RN, Bird A. Transcriptional repression by the
methyl-CpG-binding protein MeCP2 involves a histone
deacetylase complex. Nature 1998;393:386-9.
120 Hendrich B, Bird A. Identification and characterisation of
a family of mammalian methyl-CpG-binding proteins. Mol
Cell Biol 1998;18:6538-47.
121 Ng HH, Zhang Y, Hendrich B, Johnson CA, Turner BM,
Erdjument-Bromage H, Tempst P, Reinberg D, Bird A.
MBD2 is a transcriptional repressor belonging to the
MeCP1 histone deacetylase complex. Nat Genet 1999;23:
58-61.
122 Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F,
WolV e AP. Mi-2 complex couples DNA methylation to
chromatin remodelling and histone deacetylation. Nat
Genet 1999;23:62-6.
123 Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The
thymine glycosylase MBD4 can bind to the product of
deamination at methylated CpG sites. Nature 1999;401:
301-4.
124 Bader S, Walker M, Hendrich B, Bird A, Bird C, Hooper
M, Wyllie A. Somatic frameshift mutations in the MBD4
gene of sporadic colon cancers with mismatch repair
deficiency. Oncogene 1999;18:8044-7.
125 Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke
U, Zoghbi HY. Rett syndrome is caused by mutations in
X-linked MECP2, encoding methyl-CpG-binding protein
2. Nat Genet 1999;23:185-8.
126 Hagberg B, Aicardi J, Dias K, Ramos O. A progressive syn-
drome of autism, dementia, ataxia and loss of purposeful
hand use in girls: Rett’s syndrome: report of 35 cases. Ann
Neurol 1983;14:471-9.
127 Wan M, Lee S, Zhang X, Houwink-Manville I, Song H,
Amir R, Budden S, Naidu S, Pereira JL, Lo IF, Zoghbi HY,
Schanen NC, Francke U. Rett syndrome and beyond:
recurrent spontaneous and familial MECP2 mutations at
CpG hotspots. Am J Hum Genet 1999;65:1520-9.
128 Varga-Weisz PD, Becker PB. Chromatin-remodelling
factors: machines that regulate? Curr Opin Cell Biol
1998;10:346-53.
914 Johnson
www.jmedgenet.com
129 Tyler JK, Kadonaga JT. The “dark side” of chromatin
remodelling: repressive eVects on transcription. Cell
1999;99:443-6.
130 Airio A, Pukkala E, Isomaki H. Elevated cancer incidence
in patients with dermatomyositis: a population based study.
J Rheumatol 1995;22:1300-3.
131 Shorr AF, Yacavone M, Seguin S, Jackson LW, Dennis GJ.
Dermatomyositis and malignant melanoma. Am J Med Sci
1997;313:249-51.
132 Aasland R, Gibson TJ, Stewart AF. The PHD finger:
implications for chromatin-mediated transcriptional regu-
lation. Trends Biochem Sci 1995;20:56-9.
133 Ma Q, Alder H, Nelson KK, Chatterjee D, Gu Y,
Nakamura T, Canaani E, Croce CM, Siracusa LD,
Buchberg AM. Analysis of the murine All-1 gene reveals
conserved domains with human ALL-1 and identifies a
motif shared with DNA methyltransferases. Proc Natl Acad
Sci USA 1993;90:6350-4.
134 Gibbons RJ, Bachoo S, Picketts DJ, Aftimos S, Asenbauer
B, BergoVen J, Berry SA, Dahl N, Fryer A, Keppler K,
Kurosawa K, Levin ML, Masuno M, Neri G, Pierpont ME,
Slaney SF, Higgs DR. Mutations in transcriptional regula-
tor ATRX establish the functional significance of a
PHD-like domain. Nat Genet 1997;17:146-8.
135 Chudley AE, Lowry RB. X linked alpha thalassaemia/
mental retardation (ATR-X) syndrome. J Med Genet 1992;
29:357.
136 Gibbons RJ, Picketts DJ, Villard L, Higgs DR. Mutations
in a putative global transcriptional regulator cause X-linked
mental retardation with alpha-thalassemia (ATR-X syn-
drome). Cell 1995;80:837-45.
137 Gibbons RJ, McDowell TL, Raman S, O’Rourke DM,
Garrick D, Ayyub H, Higgs DR. Mutations in ATRX,
encoding a SWI/SNF-like protein, cause diverse changes in
the pattern of DNA methylation. Nat Genet 2000;24:368-
71.
138 Muchardt C, Yaniv M. A human homologue of Saccharo-
myces cerevisae SNF2/SWI2 and Drosophila brm genes
potentiates transcriptional activation by the glucocorticoid
receptor. EMBO J 1993;12:4279-90.
139 Kingston RE, Bunker CA, Imbalzano AN. Repression and
activation by multiprotein complexes that alter chromatin
structure. Genes Dev 1996;10:905-20.
140 Muchardt C, Yaniv M. The mammalian SWI/SNF
complex and the control of cell growth. Semin Cell Develop
Biol 1999;10:189-95.
141 Versteege I, Sevenet N, Lange J, Rousseau-Merck MF,
Ambos P, Handgretinger R, Aurias A, Delattre O. Truncat-
ing mutations of hSNF5/INI1 in aggressive paediatric can-
cer. Nature 1998;394:203-6.
142 Grand F, Kulkarni S, Chase A, Goldman JM, Gordon M,
Cross NC. Frequent deletion of hSNF5/INI1, a compo-
nent of the SWI/SNF complex, in chromic myeloid leuke-
mia. Cancer Res 1999;59:3870-4.
143 Pirrotta P. Pc-G complexes and chromatin silencing. Curr
Opin Genet Dev 1997;7:249-58.
144 Yu BD, Hess JL, Horning SE, Brown GA, Korsmeyer SJ.
Altered Hox expression and segmental identity in Mll-
mutant mice. Nature 1995;378:505-8.
145 Thirman MJ, Gill HJ, Burnett RC, Mbangkollo D,
McCabe NR, Kobayashi H, Ziemin-van der Poel S,
Kaneko Y, Morgan R, Sandberg AA, Chaganti RS, Larson
RA, Le Beau MM, Diaz MO, Rowley JD. Rearrangement
of the MLL gene in acute lymphoblastic and acute myeloid
leukaemias with 11q23 chromosomal translocations. N
Engl J Med 1993;329:909-14.
146 Rozenblatt-Rosen O, Rozovskaia T, Burakov D, Sedkov Y,
Tillib S, Blechman J, Nakamura T, Croce CM, Mazo A,
Canaani E. The C-terminal SET domains of ALL-1 and
Trithorax interact with the INI1 and SNR1 proteins, com-
ponents of the SWI/SNF complex. Proc Natl Acad Sci USA
1998;95:4152-7.
147 Faust C, Schumacher A, Holdener B, Magnuson T. The
eed mutation disrupts anterior mesoderm production in
mice. Development 1995;121:273-85.
148 van der Vlag J, Otte AP. Transcriptional repression
mediated by the human polycomb-group protein EED
involves histone deacetylation. Nat Genet 1999;23:474-8.
149 Turner BM. Histone acetylation and epigenetic code.
BioEssays (in press).
150 Imai SI, Armstrong CM, Kaeberlein M, Guarente L.
Transcriptional silencing and longevity protein Sir2 is an
NAD-dependent histone deacetylase. Nature 2000;403:
795-800.
151 Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pil-
lus L, Sternglanz R. The silencing protein SIR2 and its
homologues are NAD-dependent protein deacetylases.
Proc Natl Acad Sci USA 2000;97:5807-11.
152 Sherman JM, Pillus L. An uncertain silence. Trends Genet
1997;13:308-13.
153 Guarente L. Sir2 links chromatin silencing, metabolism
and aging. Genes Dev 2000;14:1021-6.
Chromatin modification and disease 915
www.jmedgenet.com
