
Letter
Relics of repeat-induced point mutation direct
heterochromatin formation in Neurospora crassa
Zachary A. Lewis,
1
Shinji Honda,
1
Tamir K. Khlafallah,
1
Jennifer K. Jeffress,
1
Michael Freitag,
2
Fabio Mohn,
3
Dirk Schu
¨
beler,
3
and Eric U. Selker
1,4
1
Institute of Molecular Biology, University of Oregon, Eugene, Oregon, 97403-1229, USA;
2
Center for Genome Research and
Biocomputing, Department of Biochemistry and Biophysics, Oregon State University, Corvallis, Oregon 97331, USA;
3
Friedrich
Miescher Institute for Biomedical Research, 4058 Basel, Switzerland
Both RNAi-dependent and -independent mechanisms have been implicated in the establishment of heterochromatin
domains, which may be stabilized by feedback loops involving chromatin proteins and modifications of histones and
DNA. Neurospora crassa sports features of heterochromatin found in higher eukaryotes, namely cytosine methylation
(5mC), methylation of histone H3 lysine 9 (H3K9me), and heterochromatin protein 1 (HP1), and is a model to investigate
heterochromatin establishment and maintenance. We mapped the distribution of HP1, 5mC, H3K9me3, and H3K4me2 at
100 bp resolution and explored their interplay. HP1, H3K9me3, and 5mC were extensively co-localized and defined 44
heterochromatic domains on linkage group VII, all relics of repeat-induced point mutation. Interestingly, the centromere
was found in an ;350 kb heterochromatic domain with no detectable H3K4me2. 5mC was not found in genes, in contrast
to the situation in plants and animals. H3K9me3 is required for HP1 localization and DNA methylation in N. crassa.In
contrast, we found that localization of H3K9me3 was independent of 5mC or HP1 at virtually all heterochromatin regions.
In addition, we observed complete restoration of DNA methylation patterns after depletion and reintroduction of the
H3K9 methylation machinery. These data show that A:T-rich RIP’d DNA efficiently directs methylation of H3K9, which
in turn, directs methylation of associated cytosines.
[Supplemental material is available online at www.genome.org. The microarray data reported in this work have been
submitted to Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) under accession no. GSE12690.]
Chromatin is the relevant substrate for all DNA-mediated pro-
cesses in eukaryotes. Arrays of nucleosomes consisting of ;146 bp
of DNA wrapped around an octamer of four histone proteins (H3,
H4, H2A, and H2B) represent the lowest level of chromatin orga-
nization. Interactions of nucleosomes with each other and with
nonhistone chromatin proteins and RNAs are thought to mediate
functionally important higher-order chromatin structures. Gene-
rich ‘‘euchromatin’’ exists in an open conformation during much
of the cell cycle, facilitating DNA transactions such as transcrip-
tion. In contrast, the densely staining ‘‘heterochromatin’’ remains
highly condensed throughout the cell cycle, exhibits low levels of
transcription, and contains relatively few genes (Luger 2006;
Grewal and Jia 2007).
The core histones are subject to extensive covalent mod-
ifications (e.g., by phosphorylation, acetylation, methylation, and
ubiquitination) that can impact chromatin structure by pro-
moting or inhibiting nucleosome interactions, or by serving as
binding sites for proteins or protein complexes such as chromatin
remodelers. Generally, euchromatin is enriched for methylated
H3K4 within active genes and is rich in acetylated histones. Some
histone modifications in euchromatin are essential for transcrip-
tional memory during development or for mounting appropriate
and timely transcriptional responses to environmental stimuli.
Conversely, heterochromatin is typically hypoacetylated and
enriched for methylated H3K9 (Bhaumik et al. 2007). Though
generally transcriptionally silent, heterochromatin is essential for
proper centromere function and promotes genome stability by
preventing illegitimate recombination between repeated DNA
(Grewal and Jia 2007; Peng and Karpen 2008).
In addition to modification of histones, many organisms
methylate some cytosines in DNA. Such DNA methylation in
eukaryotes plays roles in development, genomic imprinting, X-
chromosome inactivation, silencing of transposons, and gene
regulation (Miura et al. 2001; Reik and Walter 2001; Reik et al. 2001;
Heard and Disteche 2006; Weber and Schubeler 2007). In animal
genomes, 5mC is restricted to CpG dinucleotides whereas in plants
and some fungi (e.g., Neurospora crassa), DNA methylation occurs
in both symmetric (CpG, CpHpG; H = A, C, or T) and asymmetric
contexts (CHH) (Suzuki and Bird 2008). In plants and animals,
hemimethylated symmetrical sites produced during replication are
recognized and methylated to propagate methylation patterns
(Saze et al. 2003; Bostick et al. 2007; Sharif et al. 2007).
The mechanisms that contribute to the maintenance of
asymmetric DNA methylation and those that establish DNA
methylation remain ill-defined; however, in some cases it is clear
that histone methylation directs DNA methylation. The first, and
most clear-cut example came from studies with the filamentous
fungus N. crassa, which showed that tri-methylation of H3K9 by
the lysine methyltransferase (KMT) defective in methylation-5
(DIM-5) is essential for DNA methylation (Tamaru and Selker
2001). The N. crassa homolog of heterochromatin protein 1 (HP1),
which binds methylated H3K9, is also essential for DNA methyl-
ation (Freitag et al. 2004a). HP1 recruits DIM-2, the DNA meth-
yltransferase (DNMT) responsible for all 5mC in vegetative cells
(Kouzminova and Selker 2001; Honda and Selker 2008). Similarly,
H3K9 methylation is important for some DNA methylation in
both plants and animals (Jackson et al. 2002; Lehnertz et al. 2003).
In addition, plants carry out RNA-directed DNA methylation,
4
Corresponding author.
Article published online before print. Article and publication date are at
http://www.genome.org/cgi/doi/10.1101/gr.086231.108.
19:427–437 Ó 2009 by Cold Spring Harbor Laboratory Press; ISSN 1088-9051/09; www.genome.org Genome Research 427
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from

a process that involves the DNMT DRM2 and components of the
RNAi machinery (Henderson and Jacobsen 2007). Components of
the RNAi pathway are dispensable for DNA methylation in N.
crassa (Freitag et al. 2004b).
To develop a more complete understanding of how 5mC is
controlled, it is essential to determine the genomic location of this
modification. In plants, Methylated DNA ImmunoPrecipitation
(MeDIP) coupled with microarray analysis (Zhang et al. 2006;
Zilberman et al. 2007), and more recently whole genome shotgun
sequencing of bisulfite-treated DNA (Cokus et al. 2008; Lister et al.
2008), revealed dense methylation of repeated sequences and
transposons that are highly concentrated within the pericentric
heterochromatin domains. Methylation was also found in the
coding regions of genes, but not in the promoters, of over 30% of
expressed genes, indicating that transcriptional repression is not
necessarily an outcome of DNA methylation. In contrast, most
CpG dinucleotides within the mammalian genome are methyl-
ated, though many promoters contain C:G rich ‘‘CpG islands’’
that lack DNA methylation (Suzuki and Bird 2008).
Isolation and characterization of a fraction of the methylated
DNA from N. crassa suggested that most methylated sequences are
relics of repeat-induced point (RIP) mutation, a premeiotic genome
defense system that results in C to T changes within duplicated
sequences (Selker et al. 2003). Moreover, introduction of RIP’d or
A:T rich DNA into N. crassa triggered DNA methylation de novo,
suggesting that positive signals target methylation to specific DNA
sequences (Selker and Stevens 1987; Miao et al. 2000; Tamaru and
Selker 2003). Interestingly, lightly RIP’d sequences were unable to
trigger de novo methylation, but were able to maintain previously
established methylation (Singer et al. 1995; Selker et al. 2002). This
revealed that N. crassa has the capacity to perform both de novo and
maintenance methylation. Still, the extent to which each pathway
contributes to total methylation levels is not clear.
Since chromatin modifications impact DNA methylation, it
is important to determine what other features comprise the
chromatin environments marked by 5mC. Analysis of H3K9me3
and HP1 localization at a small number of RIP’d regions suggested
that these components are co-localized with 5mC in N. crassa
(Tamaru et al. 2003; Honda and Selker 2008), but the extent of co-
localization throughout the genome is unknown. Recent work in
mammalian cells revealed a strong inverse correlation between
H3K4 methylation and 5mC (Weber et al. 2007; Meissner et al.
2008). Despite advances, much remains to be learned about the
chromosomal contexts that promote or inhibit DNA methylation.
Knowledge of the distribution of chromatin marks should
also help elucidate interrelationships between them. Positive
feedback loops have already been implicated in establishment and
maintenance of heterochromatin domains in several organisms.
In Schizosaccharomyces pombe, domains of H3K9 methylation are
found at centromeres, telomeres, and the silent mating type loci
(Cam et al. 2005). Maintenance of heterochromatin domains in
this yeast involves the H3K9 binding protein Swi6 (homolog of
HP1), which is essential for the spread of H3K9 methylation be-
yond RNAi-dependent nucleation sites (Hall et al. 2002). In addi-
tion, the Clr4 H3K9 KMT binds methylated H3K9 via its chromo
domain, thereby facilitating methylation of adjacent nucleosomes
(Zhang et al. 2008). Similarly, Drosophila and mammalian
SU(VAR)3-9 H3K9 KMTs interact with the methyl H3K9 binding
protein HP1 (Aagaard et al. 1999; Schotta et al. 2002). In Arabi-
dopsis thaliana, the chromo domain of the CMT3 DNMT interacts
with H3 methylated at K9 and K27 to target 5methyl-cytosine
(5mC) and KRYPTONITE (also known as SUVH4), an H3K9 KMT,
binds 5mC via its SRA domain to target K9 methylation (Lindroth
et al. 2004; Johnson et al. 2007).
We mapped the chromosomal distribution of methylated
H3K9, HP1, and 5mC and found that these co-localize to form
discrete heterochromatin domains within the N. crassa genome.
We also found that while positive feedback occurs within the DNA
methylation pathway, it is not required for maintenance of most
heterochromatin domains in Neurospora. Finally, we determined
that relics of repeat-induced point mutation trigger efficient de
novo H3K9 and cytosine methylation.
Results
Distribution of 5mC in N. crassa
To investigate heterochromatin organization within the N. crassa
genome, we mapped the distribution of 5mC, H3K9me3, and HP1
across an entire chromosome. The N. crassa genome is composed of
seven chromosomes covering ;42 Mb. We chose LGVII for these
analyses because its current chromosome assembly contains few gaps,
covers ;10 % of the genome, and optical mapping data for LGVI I
suggest that the current genome assembly covers almost the entire
genetically mapped centromere (Centola and Carbon 1994) (http://
www.broad.mit.edu /anno tatio n/g enome/neurospora/Home.html).
We designed a custom microarray containing oligonucleotide
probes covering N. crassa LGVII and 37 previously identified
methylated regions (representing regions from all seven chromo-
somes) at 100 bp resolution and determined the distribution of 5
mC in wild-type N. crassa strain 74-OR-23IV by MeDIP coupled
with microarray analysis (Weber et al. 2005). Because most ‘‘re-
peated’’ sequences in the N. crassa genome have been subjected to
RIP, which typically causes 10%–20% sequence divergence, virtu-
ally all of our oligos were unique sequences (see Methods). In-
spection of the MeDIP data revealed that DNA from all 37
previously identified methylated regions was enriched in the
immunoprecipitated fraction, validating our experimental ap-
proach (Fig. 1). We showed previously that these methylated
sequences have been altered by RIP (Selker et al. 2003). The RIP
machinery, which produces C to T mutations, exhibits a preference
for CpA dinucleotides. Indeed, analyses of the frequencies of TpA
and CpA, relative to the frequencies of control dinucleotides, can
identify sequences that have been subjected to RIP. Typically,
sequences that have not been subjected to RIP exhibit values less
than 0.8 for the ‘‘RIP product index’’ (TpA / ApT) and values greater
than 1.1 for the ‘‘RIP substrate index’’ (CpA + TpG/ ApC + GpT). In
contrast, RIP’d regions typically exhibit values greater than 1.1 and
less than 0.9 for RIP product and substrate indices, respectively
(Margolin et al. 1998; Selker et al. 2003). A composite RIP index
(CRI) can be determined by subtractingthe substrate index from the
product index; thus, a positive CRI value implies that the DNA has
been subjected to RIP. We plotted the %GC and the CRI across each
previously identified region. 5mC was enriched throughout the
RIP’d regions and did not extend significantly into unRIP’d DNA.
We next examined the distribution of 5mC across N. crassa
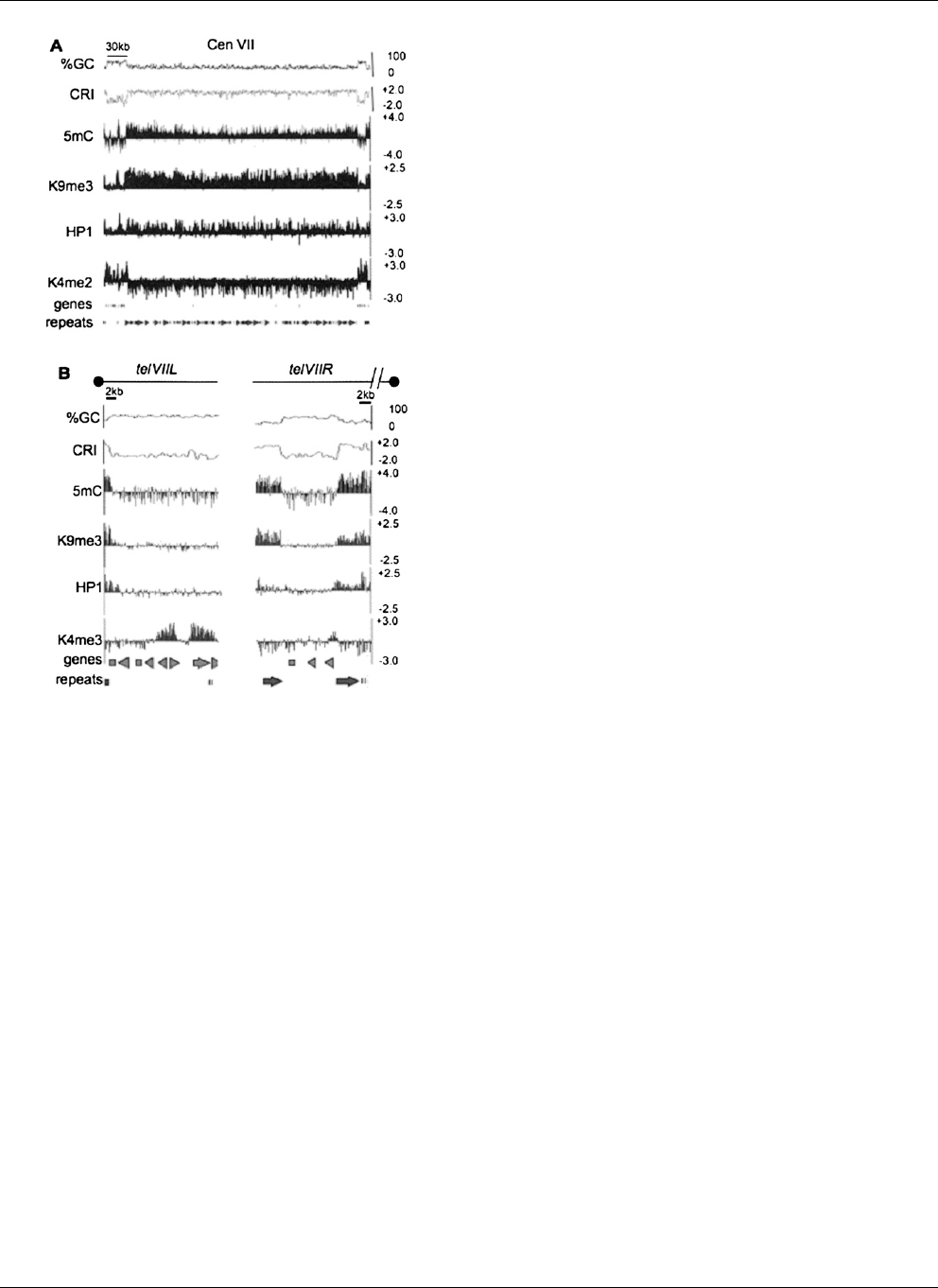
LGVII (Fig. 2). Interestingly, a large region (;346 kb) comprising
the putative pericentric/centromeric DNA was enriched in the
MeDIP fraction (Fig. 3A). In addition, we observed numerous
methylated domains along both chromosome arms including
short methylated subtelomeric domains (Fig. 3B). To determine
the location of 5mC in an unbiased fashion, peaks of enrichment
were identified using ChIPOTle (
Chromatin Immuno Pre-
cipitation On Tiled microarrays) ChIP-chip data analysis software
428 Genome Research
www.genome.org
Lewis et al.
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from
(Buck et al. 2005). Forty-six peaks were identified by ChIPOTle on
LGVII ranging in size from 0.6 to 138.5 kb (Supplemental Table 1).
Consistent with visual inspection of the data, peaks 29, 30, and 31
covered virtually all 346 kb of the putative pericentromere/cen-
tromere. The remaining 43 peaks were distributed on both arms of
LGVII and ranged in size from 0.6 to 21.4 kb (average of 7.1 kb
excluding centromeric region). Overall, methylated regions on
LGVII included 652 kb, or 16.5% of the 3.9 Mb chromosome. This
is somewhat higher than expected since previous studies indicated
that ;2% of cytosines are methylated in N. crassa (Foss et al.
1993), but is consistent with previous reports of heterogeneous
methylation within N. crassa cultures (Selker et al. 1993).
Co-localization of 5mC and H3K9me3
H3K9 methylation in wild-type N. crassa exists predominantly, if
not exclusively, as tri-methyl K9 (Tamaru et al. 2003) and this
chromatin modification has been shown to co-localize with 5mC
at several loci (Honda and Selker 2008). We determined the extent
of co-localization by examining the distribution of H3K9me3 us-
ing our high-resolution tiled microarray. Enrichment of H3K9me3
was found within all regions that had been previously examined,
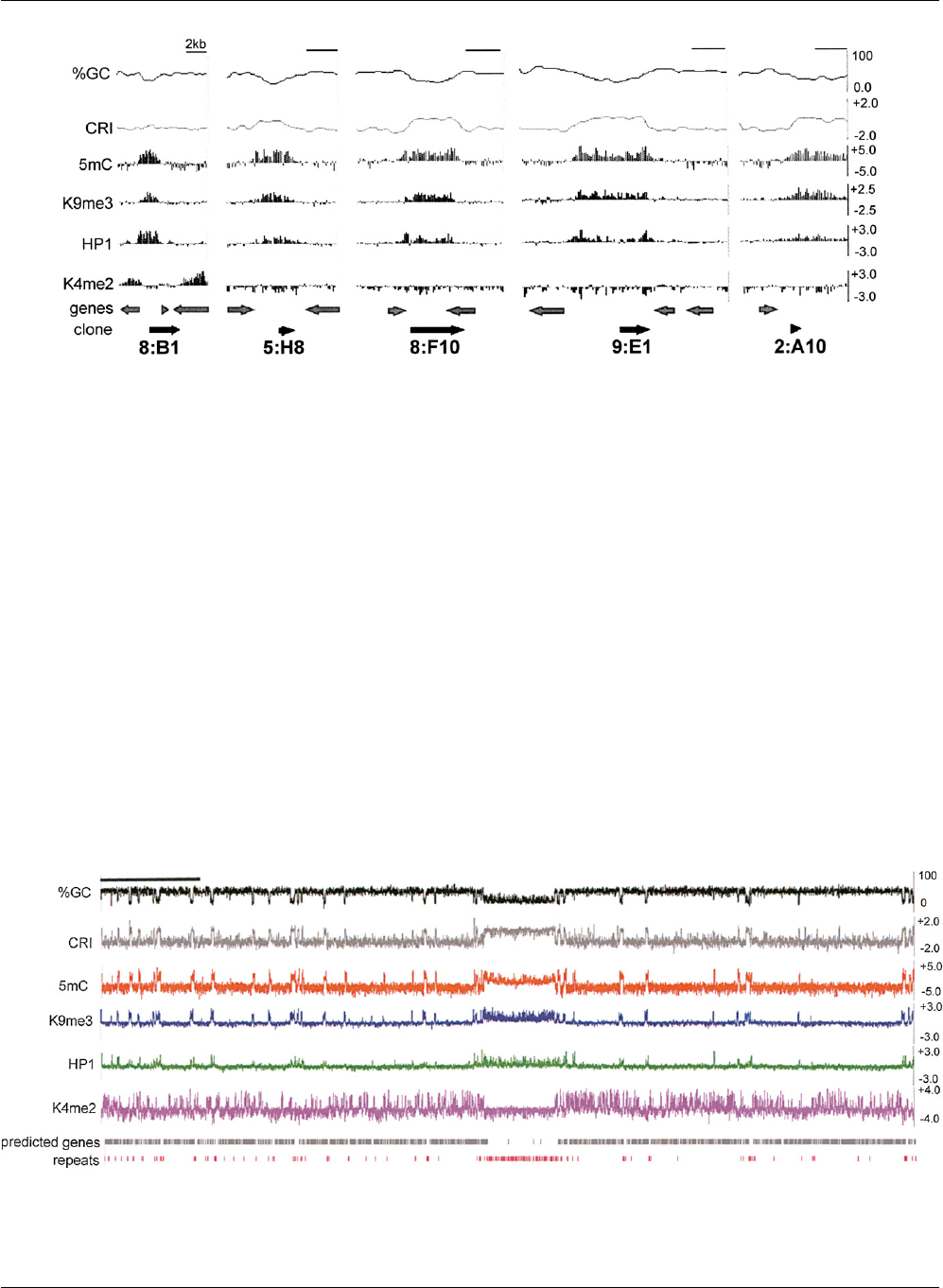
once again validating the experimental approach (8:B1, 5:H8,
8:F10; Fig. 1). Importantly, experiments using two different anti-
bodies that recognize H3K9me3 yielded virtually identical results
(see Methods). Visual inspection of the data revealed striking co-
localization of 5mC and H3K9me3 across LGVII (Fig. 2). As with
5mC, H3K9me3 was enriched within the putative centromeric
region (Fig. 3A) and in dispersed regions along both arms, in-
cluding subtelomeric domains (Fig. 3B). We used ChIPOTle to
identify peaks of H3K9me3 enrichment; 45 peaks were identified
ranging in size from 0.5 to 151.1 kb (Supplemental Table 2). Peaks
27–31 covered virtually all 346 kb of the putative centromere,
whereas the remaining 40 peaks were distributed on both arms of
Figure 2. Chromatin modification profile for N. crassa LGVII. Base composition of the 3.9 Mb LGVII is shown as the moving average of %GC and the
CRI calculated for 500 bp windows with 100 bp steps at the top of the plot. Enrichment values for MeDIP and ChIP-chip experiments are shown as log
2
values indicated on the y-axis (right) for immunoprecipitation experiments with antibodies to 5mC, H3K9me3, green fluorescent protein for HP1-GFP
(HP1) and H3K4me2. The positions of predicted open reading frames (predicted genes) and repeats are indicated below. The scale bar on the top left
indicates 0.5 Mb.
Figure 1. Chromatin modification profile of previously identified methylated regions. Data from five representative control regions are shown. A scale
bar indicating 2 kb is shown at the top right of each plot. The base composition (%GC) and a composite RIP index (CRI) of each region, plotted as the
moving average for 500 bp windows with 100 bp steps, are shown at the top of each plot. Enrichment values for MeDIP and ChIP-chip experiments are
shown as log
2
values (y-axis) for immunoprecipitation experiments with antibodies to 5mC, H3K9me3, green fluorescent protein for HP1-GFP (HP1) and
H3K4me2 for each region. The positions of predicted open reading frames (genes) and the previously identified methylated DNA clones, with their
identification numbers, are shown at the bottom.
Heterochromatin in N. crassa
Genome Research 429
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from
LGVII, ranging in size from 0.5 to 21.6 kb. H3K9me3 regions in-
cluded 646 kb, or 16.4% of the 3.9-Mb chromosome, close to that
detected by MeDIP (see above).
We examined the degree of overlap between the 5mC peaks
and the H3K9me3 peaks. Of the 46 5mC peaks, 41 of these co-
incided with an H3K9me3 peak (Supplemental Table 1). It is likely
that the five 5mC regions that did not overlap with H3K9me3
peaks contain low levels of the modified histone that were not
detected by ChIPOTle. Indeed, three of these 5mC regions (peaks
10, 27, and 44; Supplemental Table 1) appeared to contain
H3K9me3 by visual inspection and were identified as H3K9me3
peaks in one experiment, suggesting that low levels of H3K9me3
are present in these 5mC regions (data not shown). The fourth
peak that did not overlap with an H3K9me3 peak displayed ob-
vious enrichment of HP1 and appeared to have low levels of
H3K9me3 by visual inspection and conventional ChIP (peak 34;
Fig. 4; Supplemental Table 1; see below). The fifth peak that did
not overlap with H3K9me3 had low levels of 5mC enrichment and
may contain H3K9me3 that was not detectable under our exper-
imental conditions. Notably, all 45 H3K9me3 peaks coincided
with 5mC peaks (Supplemental Table 2). Taken together, these
data show that 5mC and H3K9me3 are co-localized within the
N. crassa genome.
Complex localization of HP1
HP1 interacts directly with both H3K9me3 and the DIM-2 DNMT via
its chromo domain and chromo shadow domain, respectively
(Freitag et al. 2004a; Honda and Selker 2008). These interactions are
essential for DNA methylation in N. crassa. To determine if HP1 is
present at all methylated regions and if HP1 is localized to both
methylated and unmethylated regions, we examined the distribu-
tion of an HP1-GFP fusion protein expressed from the native HP1
locus and compared its localization to that of 5mC and H3K9me3 by
ChIP-chip. HP1 was generally co-localized with these two mod-
ifications (Figs. 1–3). Surprisingly, however, variable enrichment of
HP1-GFP was detected, both among H3K9me3 regions and within
some regions. It seems unlikely that this distribution resulted from
altered protein function due to the GFP tag because the strain
expressing this fusion protein displays wild-type levels of DNA
methylation (Honda and Selker 2008). Moreover, we performed
conventional ChIP experiments at four heterochromatic regions
using a strain expressing FLAG-tagged HP1. Like HP1-GFP, the HP1-
33 FLAG fusion protein was expressed from the native HP1 locus
and this strain displayed wild-type levels of DNA methylation (S.
Honda and E.U. Selker, unpubl.). Peak 35 (Supplemental Table 1)
exhibited high levels of both H3K9me3 and HP1 throughout the
RIP’d region (Fig. 4). In contrast, HP1 exhibited significantly higher
enrichment near the edges of peak 33 (Supplemental Table 1),
whereas H3K9me3 was relatively evenly distributed (Fig. 4). Peak 34
(Supplemental Table 1) was significantly enriched for HP1 even
though H3K9me3 levels were low at this region (Fig. 4); the enrich-
ment values for H3K9me3 and HP1 were of 1.9 and 2.6, respectively.
Although theseenrichment valuesare similar, HP1 levelswere;45%
of the maximum enrichment (sixfold), while H3K9me3 levels were
less than 20% of the maximal enrichment (10-fold) suggesting that
while HP1 and H3K9me3 co-localize, they differ quantitatively.
Peak 19 had low levels of HP1 even though H3K9me3 enrichment
levels were high at this region (Fig. 4). Thus, conventional ChIP
experiments confirmed the results seen in the microarray experi-
ments. In addition, similar results were obtained for ChIP-chip
experiments using strains expressing GFP-tagged and FLAG-tagged
HP1 (data not shown), confirming that the location and amount of
HP1 binding is not a direct function of H3K9me3.
DNA sequences in heterochromatic regions are products of RIP
A plot of the CRI for LGVII revealed that heterochromatin, defined
by H3K9me3 and 5mC, is predominantly associated with RIP’d
DNA. Major peaks of 5mC and H3K9me3 enrichment coincided
with CRI peaks (Fig. 2). We next asked if all methylated DNA on
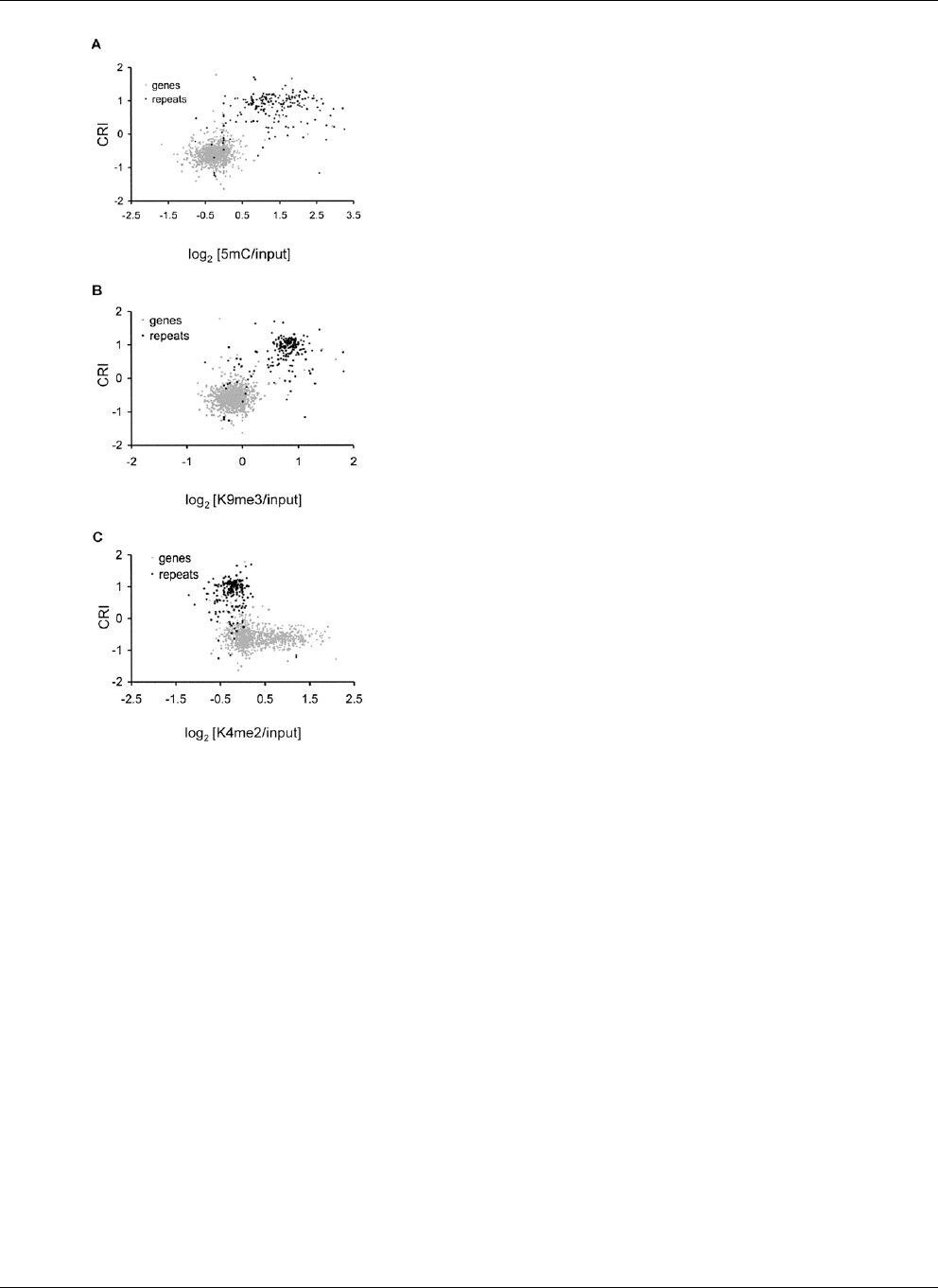
LGVII is present within relics of RIP. We calculated the CRI for all 46
methylated regions described above. The average CRI for all 46
methylated regions was 0.69 and 44 regions had a positive CRI. Both
observations suggest that methylated DNA, in general, had been
extensively RIP’d. We asked if methylated regions also had low levels
of G:C, also expected of sequences exposed to RIP. Indeed, 80% of
Figure 3. Chromatin modification profile for the LGVII centromere and
telomeres. For (A) N. crassa LGVII centromere and (B) N. crassa chromo-
some ends, nucleotide composition is shown as the moving average of
%GC and CRI calculated for 500 bp windows with 100 bp steps at the top
of the plot. Enrichment values for MeDIP and ChIP-chip experiments are
shown as log
2
values indicated on the y -axis (right) for immunoprecipi-
tation experiments using antibodies to 5mC, H3K9me3, green fluores-
cent protein for HP1-GFP (HP1) and H3K4me2. The position of predicted
open reading frames (genes) and repeats are indicated below. The scale bars
on the top indicate 30 kb and 2 kb for (A)and(B), respectively. The black
dots above each plot in (B) indicate the position of the telomere. The broken
line above telVIIR indicates that sequences containing the telomere repeats
for telVIIR are missing from the current LGVII sequence assembly.
Lewis et al.
430 Genome Research
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from

methylated regions had a G:C content of 40% or lower and the av-
erage GC contentforall 46 methylated regionswas34.4%(compared
to an average G:C content of 54.7% for predicted LGVII genes).
We next asked if all RIP’d DNA is associated with DNA
methylation. We used ChIPOTle to call peaks of RIP’d DNA,
yielding a total of 587 kb of DNA for LGVII. 96% of this predicted
RIP’d DNA overlapped with a peak of 5mC. Interestingly, peaks of
RIP’d DNA that did not overlap with 5mC exhibited one or more
of the following properties: (1) The RIP’d region was less than 300
bp in length, (2) the RIP’d region overlapped with a region that
contained H3K4me2, and/or (3) the RIP’d region had a G:C con-
tent higher than 45%. These data indicate that in N. crassa, A:T
rich, RIP’d sequences that lack H3K4me2 and are greater than 300
bp in length are targeted for H3K9me3 and 5mC.
In contrast to the A:T -rich, RIP’d sequences, N. crassa genes are
G:C-rich. We found that only 0.5% of the predicted ORFs on LGVII
(5/1008) significantly overlap (>90%)with5mCregionsandthese
appeared to be pseudogenes that resulted from RIP. They show
a positive CRI, their predicted coding regions are short (<100 amino
acids) and BLAST searches failed to identify similar sequences in the
NCBI nonredundant database. Other apparent overlap between
genes and 5mC turned out to be simply a consequence of the limited
resolution MeDIP assay, which uses DNA sheared to ;500 bp. For
example, the gene to the left of the 8:B1 region (Fig. 1) appears to
overlap with the edge of the 5mC region, but Southern hybridizations
demonstrated that it is not methylated (data not shown). Taken to-
gether, our data show that genes are not targets of DNA methylation
in Neurospora, unlike the situation in plants and animals.
DNA methylation is a widespread feature of repeated
sequences of plants and animals, and this is also true for Neuros-
pora. N. crassa repeated sequences are divergent, with numerous
C:G to T:A nucleotide changes indicative of RIP. We found that
89% of the annotated repeats on LGVII overlapped with called
5mC peaks. Figure 5 summarizes the distribution of 5mC and
H3K9me3 within LGVII genes and repeated sequences.
DNA methylation is not affected in a double
Argonaute mutant
Some DNA methylation in plants depends on RNAi machinery
(Henderson and Jacobsen 2007), but this does not appear to be the
case in N. crassa. Both maintenance and de novo DNA methyla-
tion occurred normally in a dicer double mutant, a triple RNA-
dependent RNA polymerase mutant, and two single Argonaute
mutants (Freitag et al. 2004b; Chicas et al. 2005). It remained
a formal possibility, however, that the two Argonaute proteins
have redundant roles in DNA methylation. To test this, we per-
formed a MeDIP experiment with a double Argonaute mutant and
determined the distribution of 5mC using our high-resolution
microarray. DNA methylation was detected at all methylated
regions identified in the wild-type strain (Supplemental Fig. 1).
Southern blots also revealed that DNA methylation was equivalent
to that in wild type at all the heterochromatic loci examined (data
not shown). Thus, our findings strengthen the conclusion that the
RNAi machinery is not required for DNA methylation in N. crassa.
Distribution of a euchromatic mark
In contrast to heterochromatin, actively transcribed genes are
methylated at K4 of histone H3 in eukaryotes that have been
examined (Bhaumik et al. 2007). Because levels of H3K4me2
are inversely correlated with DNA methylation in mammals
(Weber et al. 2007; Meissner et al. 2008), we performed ChIP-chip
Figure 4. Complex localization patterns of HP1. Microarray data from immunoprecipitation experiments using antibodies to 5mC, H3K9me3, green
fluorescent protein (for HP1-GFP; HP1), and H3K4me2 are shown as log
2
values for 5mC peaks 35, 33, 34, and 19 (Supplemental Table 1). The position of
predicted open reading frames (genes) and repeats are indicated below. Conventional ChIP was performed for each region using antibodies to H3K4me2,
H3K9me3, and FLAG (for HP1-FLAG) to validate the microarray data. PCR products obtained using whole cell extracts (WCE), or the indicated immu-
noprecipitate fraction as the template, were resolved by gel electrophoresis. The black bars labeled P1–P5 depict the position of the expected PCR
products within each region. The coding region of histone H4 was used as an internal euchromatin control. The labels to the right of each autoradiograph
identify specific PCR products. The enrichment of each PCR product relative to the PCR product for H4 to is shown below.
Heterochromatin in N. crassa
Genome Research 431
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from
experiments to examine the distribution of this modification in N.
crassa. Similar to the case in other organisms, H3K4me2 was
enriched in the coding regions of genes. Of 1008 predicted ORFs on
LGVII, 42.5%were enriched for H3K4me2 within the genebodies.In
contrast, 5mC enrichment peaks displayed negative log
2
values,
indicative of H3K4me2 depletion (Figs. 2, 5C). Indeed, the average
log
2
enrichment value for H3K4me2 within the 46 5mC regions was
0.61 suggesting that heterochromatin and H3K4me2 are mutually
exclusive in N. crassa. In many organisms, the centromeric core is
enriched for H3K4me2 and for a centromere-specific H3 variant
(CENPA, also known as CENH3) (Sullivan and Karpen 2004; Cam
et al. 2005). Interestingly, we did not detect any enrichment of
H3K4me2 within the centromeric core of N. crassa LGVII (Fig. 3A).
Limited positive feedback from HP1 contributes to normal
H3K9 methylation
In mammals, DNA methyl-binding domain (MBD) proteins in-
teract with H3K9 KMTs (Fujita et al. 2003; Sarraf and Stancheva
2004), whereas in plants, the SRA domain present in the KRYP-
TONITE/SUVH4 H3K9 KMT binds 5mC (Johnson et al. 2007). To
examine whether DNA methylation might direct chromatin
modification in N. crassa, we compared the distribution of
H3K9me3 in the dim-2 DNMT mutant with that in a wild-type
strain. As a negative control, we examined the distribution of
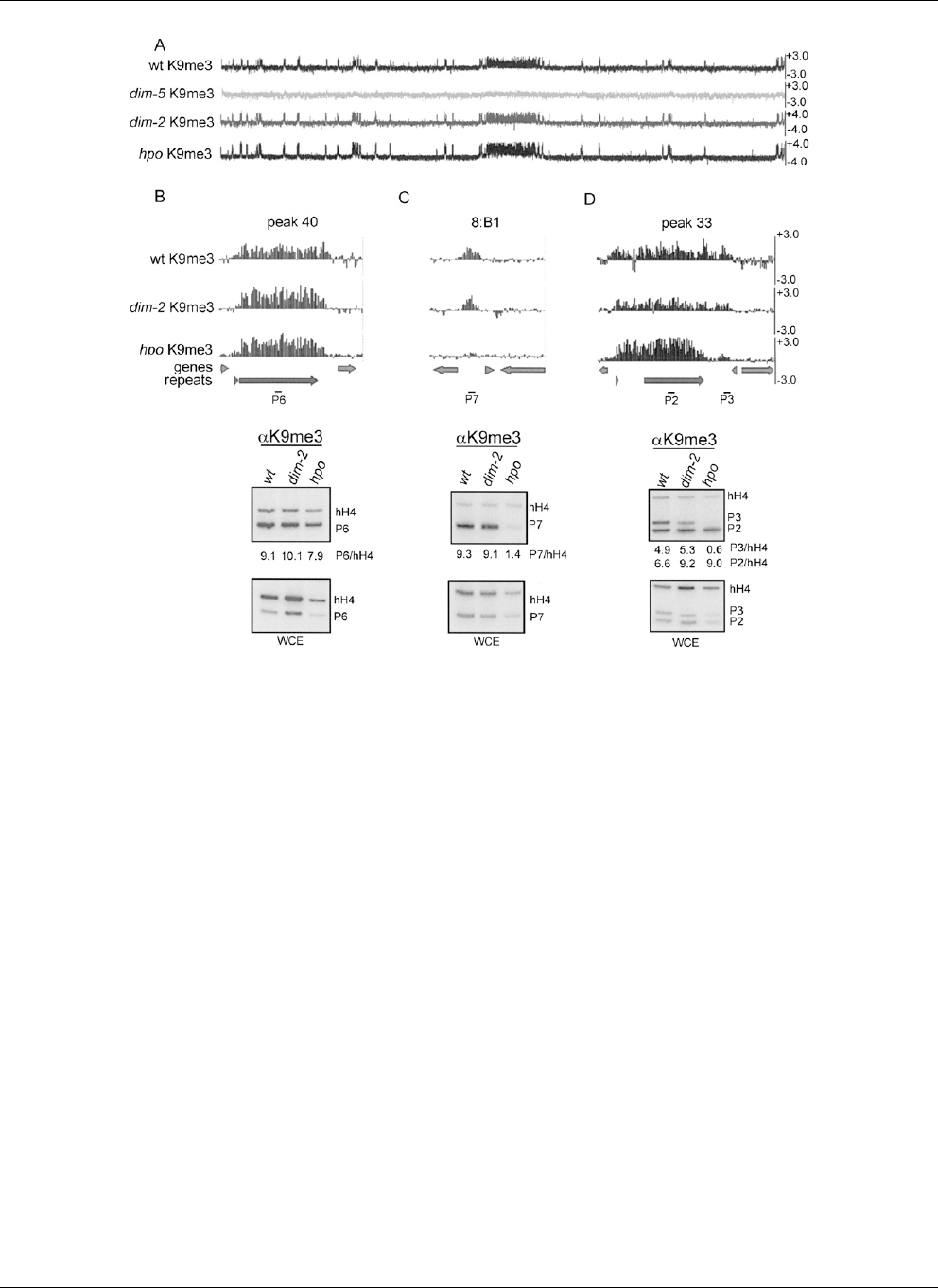
H3K9me3 in the dim-5 KMT mutant (strain N3436). We found that
H3K9me3 persists at all LGVII heterochromatic regions in the dim-2
mutant (Fig. 6). We also examined the distribution of H3K9me3 in
the hpo strain because HP1 is known to interact with numerous
proteins that modify chromatin. Most of the H3K9me3 detected in
wild-type strains was also present in the hpo mutant (Fig. 6A), but
we did identify three regions that lost H3K9me3 in the hpo mutant.
These included one region on LGVII (peak 33) and two previously
identified methylated clone sequences (8:A6 and 2:C9). HP1-
sensitive regions were short (<2.0 kbp) and had higher G:C content
than HP1-insensitive regions, although these regions had lower
G:C contents than gene sequences. The data for a typical hetero-
chromatin region (HP1-insensitive) and two of these atypical (HP1-
sensitive) regions are shown in Figure 6B. We conclude that 5mC
and HP1 are dispensable for normal H3K9me3 at most regions of
the genome but that HP1 is required for H3K9me3 at a minority of
heterochromatic regions.
Efficient de novo methylation in N. crassa
Previous studies demonstrated that both maintenance and de novo
DNA methylation occur in N. crassa (Selker et al. 2002). To assess the
fraction of methylated regions that are capable of rapid de novo
methylation, we crossed different mutant strains that lack all DNA
methylation and isolated a wild-type daughter strain. Thus, all 5mC
present in the daughter strain must be a result of de novo methyl-
ation. The distributions of 5mC from wild type, the two parental
strains (dim-2 and hpo) and the wild-type daughter strain (strain
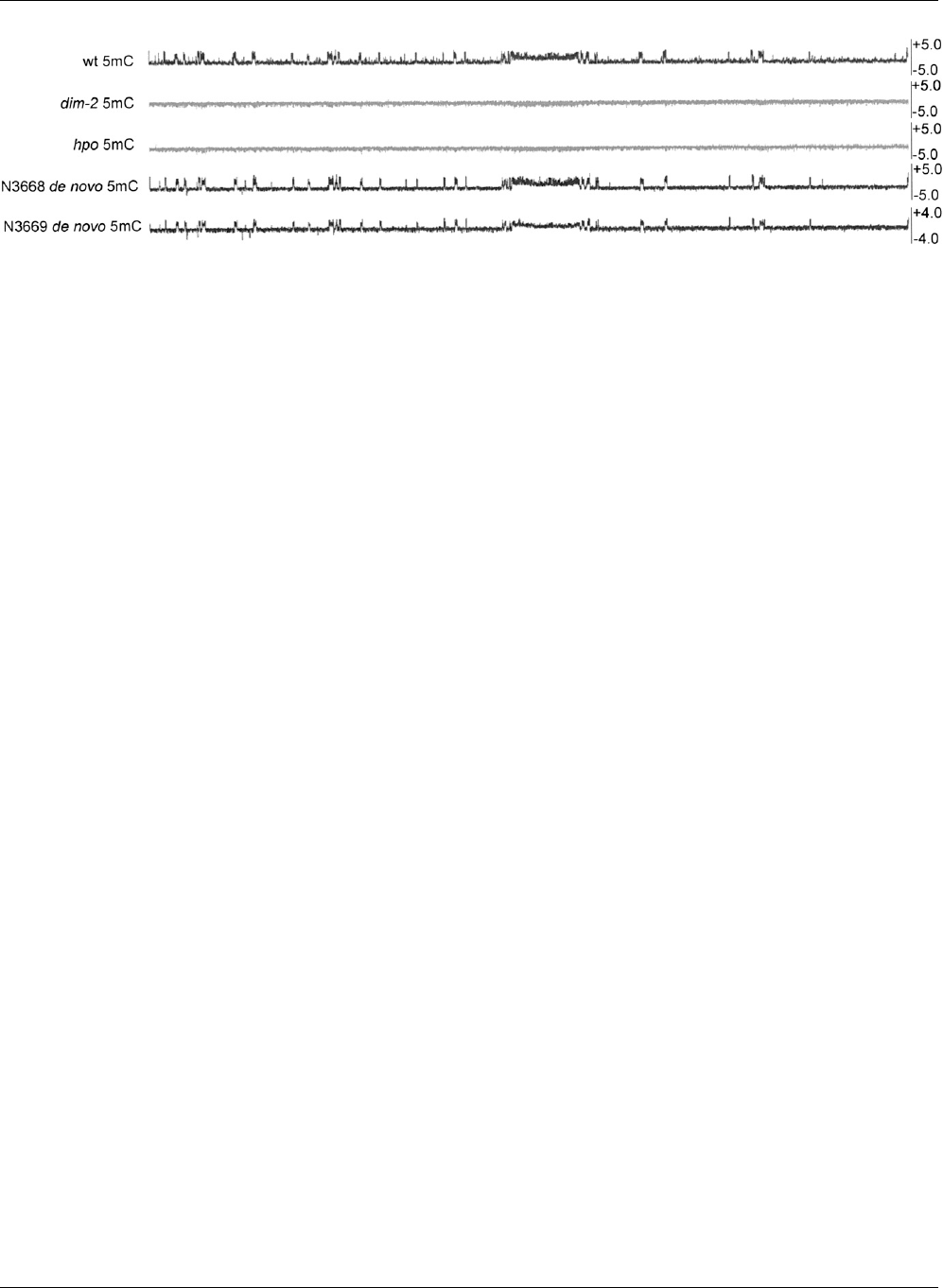
N3668) are shown for LGVII (Fig. 7). As expected, 5mC was not
detected in the two parental strains. In contrast, extensive meth-
ylation was detected in the daughter strain. Indeed, 5mC was
detected at all of the methylated regions detected in the wild-type
strain. This indicates that de novo methylation occurs rapidly in
Neurospora and that DNA methylation does not, in general,
depending on preexisting methylation.
We carried out a similar experiment to determine whether
H3K9me3 is efficiently reestablished de novo. In particular, we
crossed dim-5 (Tamaru and Selker 2001) and dim-8 (Z.A. Lewis, K.K.
Adhvaryu, S. Honda, and E.U. Selker, unpubl.) strains, which are
both defective in H3K9me3 (and 5mC), isolated a wild-type
recombinant and performed a MeDIP experiment to determine
which regions were able to undergo de novo H3K9me3 and sub-
sequent DNA methylation. Remarkably, DNA methylation was
restored at all LGVII heterochromatin regions described above.
Southern analyses were fully consistent with the MeDIP data (Sup-
plemental Fig.2).We concludethat bothH3K9me3 methylation and
DNA methylation are efficiently re-established de novo in N. crassa.
Discussion
Co-localization of 5mC and H3K9me3
We examined the detailed genomic distribution and hierarchical
relationships of DNA methylation, H3K9 methylation, and HP1 in
the filamentous fungus N. crassa. H3K9 methylation is required for
DNA methylation in this fungus and consistent with this obser-
vation, we found that the location of DNA methylation was highly
Figure 5. Genes and repeats reside in distinct chromatin environ-
ments. Scatter plots depicting the relationship between RIP and chro-
matin modifications are shown for LGVII genes (gray dots) and repeats
(black dots). The CRI value calculated for each predicted open reading
frame and annotated repeat is plotted on the y-axis. The median en-
richment value for (A) 5mC, (B) H3K9me3, and (C ) H3K4me2 for each
gene and annotated repeat is plotted on the x-axis.
Lewis et al.
432 Genome Research
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from
correlated with the location of H3K9me3. Indeed, all regions that
were enriched for H3K9me3 were also enriched for 5mC. This
suggests that H3K9 methylation is sufficient to trigger DNA
methylation in N. crassa.
Complex localization of HP1
In N. crassa, HP1 appears to directly link H3K9me3 with 5mC
(Honda and Selker 2008). Two PXVXL-related motifs in DIM-2
interact directly with the HP1 chromo shadow domain. In-
teraction of HP1 with methylated H3K9 is well established
(Bannister et al. 2001; Lachner et al. 2001). Consistent with this,
we found that HP1 was generally co-localized with H3K9 meth-
ylation at N. crassa heterochromatic domains. Interestingly, HP1
was not highly enriched at all H3K9me3 regions, however, and
HP1 enrichment was found to be high at one region that showed
low levels of H3K9me3. Furthermore, in some regions HP1 dis-
played a nonuniform distribution relative to H3K9me3 and 5mC
(Fig. 4). Several possible models could account for the complex
pattern of HP1 localization observed in our experiments. (1) HP1-
interacting proteins may contribute to the pattern of HP1 locali-
zation by recognizing additional structural features such as
histone modifications or DNA sequences. Accessory factors that
promote chromatin association of HP1 have been identified in
mammals and Drosophila (Nielsen et al. 2001; Shi et al. 2008). (2)
Additional chromo domain proteins could compete for binding of
H3K9me3 and thereby influence the distribution of HP1. Mam-
mals encode multiple HP1 proteins that show distinct localization
patterns and the fission yeast S. pombe encodes several H3K9-
binding, chromo domain proteins that each display preferential
binding at distinct genomic locations (Maison and Almouzni
2004; Grewal and Jia 2007). N. crassa encodes several chromo
domain proteins (Borkovich et al. 2004). (3) The ChIP procedure
may preferentially enrich for sites where HP1 forms a stable
complex with the chromatin fiber. Both high and low mobility
populations of HP1 exist within cells (Cheutin et al. 2004;
Schmiedeberg et al. 2004). Importantly, the nonuniform distri-
bution of HP1 relative to that of H3K9me3 (and 5mC) may in-
dicate that only transient association with chromatin is required
for HP1-directed DNA methylation by DIM-2. Alternatively, HP1
may be more uniformly distributed with H3K9me3 during a spe-
cific phase of the cell cycle, such as during replication. It will be
important to determine how a somewhat variable pattern of HP1
localization can lead to the observed pattern of DNA methylation.
Figure 6. Distribution of H3K9 methylation in wild-type, hpo, and dim-2 strains. (A) The distribution of H3K9me3 across LGVII is shown for wild-type,
dim-5, hpo, and dim- 2 strains. (B) The distribution of H3K9me3 is plotted for wild-type, hpo,anddim-2 strains for LGVII 5mC peak 40, (C ) the previously
identified methylated region 8:B1, and (D) LGVII 5mC peak 33 (Supplemental Table 1 and Selker et al. 2003). Conventional ChIP was performed using
antibodies to H3K9me3 for the indicated strains. For each strain, PCR products obtained using the immunoprecipitated fraction (H3K9me3, top)and
whole cell extract (WCE, bottom) as a template were resolved by gel electrophoresis. The black bars numbe red P2–P7 depict the position of the expected
PCR products within each region. The coding region of histone H4 (hH4) was used as an internal euchromatic control. The labels to the right of each
autoradiogram indicate specific PCR products. The relative enrichment of each PCR product is shown below.
Heterochromatin in N. crassa
Genome Research 433
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from
RIP and DNA methylation
Previous studies revealed a close association between the process
of RIP and DNA methylation (Selker et al. 1993, 2003). The
remaining cytosines within a duplicated sequence are typically
methylated following RIP and isolation of methylated DNA
revealed that most methylated sequences appear to be relics of RIP.
Our results strongly support and extend prior evidence of a tight
connection between RIP and DNA methylation. In this study, we
have shown that both 5mC and H3K9me3 are exclusively asso-
ciated with relics of RIP all along LGVII and in other previously
identified methylated regions.
Structure of LGVII centromere and telomeres
The most notable chromosomal domain of RIP’d DNA includes
the genetically mapped centromere. In S. pombe, Drosophila, and
humans, the centromeric core domain is comprised of CENPA
containing nucleosomes interspersed with nucleosomes that
contain H3K4me2 (Sullivan and Karpen 2004; Cam et al. 2005).
In contrast, pericentric domains in these organisms typically
contain H3 methylated at K9, and in plants and animals, peri-
centric sequences contain 5mC (Amor et al. 2004; Suzuki and Bird
2008). Recent work in S. pombe revealed that heterochromatic
modifications within the pericentric domains are required for
CENP-A deposition (Folco et al. 2008). Interestingly, we failed to
identify any H3K4 methylation within the putative LGVII cen-
tromere. Moreover, 5mC and H3K9me3 were distributed broadly
throughout the regions believed to comprise the centromeric core
suggesting that N. crassa possesses a novel centromere organiza-
tion. Although it is conceivable that the current genome sequence
annotation lacks the bona fide LGVII centromeric core sequence,
this is unlikely since ChIP-sequencing experiments with the N.
crassa CENP-A protein show enrichment of the H3 variant in
sequences included on our microarray (K.M. Smith and M. Freitag,
unpubl.). Thus, our data indicate that enrichment of H3K4me2
within the centromeric core may not be a general feature of
eukaryotic centromeres. Future work to determine the distribution
of N. crassa centromere proteins should provide additional insight
into centromere organization in N. crassa.
Silencing of genes that reside near telomeres, termed the
‘‘telomere position effect,’’ has been attributed to repressive
chromatin that spreads from telomere-associated sequences into
adjacent subtelomeric chromatin (Perrod and Gasser 2003). In S.
pombe, a cenH-like repeat near telomere IL is believed to nucleate
a nearly 40 kb telomeric heterochromatin domain containing
H3K9 methylation and Swi6/HP1 (Cam et al. 2005). Similarly, we
observed localization of H3K9me3 and HP1, as well as 5mC, at the
chromosome ends in N. crassa. In contrast to the extended
domains of subtelomeric heterochromatin in S. pombe, the N.
crassa, LGVIIL 5mC/H3K9me3 telomeric domain was short and
highly correlated with A:T rich sequences. These data suggest that
heterochromatin does not spread beyond the short RIP’d
sequences found at telomeres in N. crassa (Wu et al. 2008).
Moreover, genes that are located close to subtelomeric hetero-
chromatin were found enriched for H3K4 methylation suggesting
that gene expression is not silenced in these regions.
5mC is not found in N. crassa genes
In addition to centromeres and telomeres, H3K9me3, HP1, and
5mC mapped to over 40 dispersed sites across LGVII that also show
hallmarks of RIP. None of the heterochromatic domains on LGVII
appear to contain functional genes. Conversely, coding sequences
and virtually all promoters were found to be free of 5mC, H3K9me3,
and HP1. Thus, our data do not support an extensive role for DNA
methylation, H3K9 methylation, or HP1 in regulation of tran-
scription under standard growth conditions. It is also notable that
H3K4 methylation was found in several heterochromatin-adjacent
genes (see Fig. 4), suggesting that heterochromatic domains do not
necessarily suppress expression of flanking genes. This observation
is consistent with transcript profiling experiments that do not
support a role for 5mC in regulating gene expression in N. crassa (A.
Shiver, Z.A. Lewis, and E.U. Selker, unpubl.). In plants and animals,
DNA methylation within gene bodies has been proposed to prevent
initiation at cry ptic promoters, a function apparently relegated to
methylation of H3K36 in the yeast Saccharomyces cerevisiae (Carrozza
et al. 2005; Zilberman et al. 2007). N. crassa does not appear to rely
on DNA methylation to prevent such aberrant transcription initi-
ation (Rountree and Selker 1997), but does perform H3K36 meth-
ylation (Adhvaryu et al. 2005). Perhaps gene body methylation is
important for inhibiting aberrant transcription initiation in
organisms with larger coding sequences, while H3K36 methylation
may be sufficient to provide this function in organisms with shorter
genes.
Limited feedback within the N. crassa 5mC pathway
Although we found that DNA methylation was dispensable for
H3K9 methylation at all RIP’d regions examined, it is possible that
Figure 7. N. crassa performs efficient de novo methylation of heterochromatic regions. The distribution of 5mC is shown across LGV II for wild type
(wt), the methylation deficient strains dim-2 and hpo, and wild-type daughter strains from two crosses between methylation deficient strains. Enrichment
values for MeDIP experiments are shown as log
2
values on the y-axis. The wild-type strain N3668 was obtained by crossing two strains that lack 5mC but
retain H3K9me3, dim-2,andhpo. The wild-type strain N3669 was obtained by crossing two strains that lack both H3K9me3 and 5mC, dim-5,anddim- 8.
Thus, all methylation in these strains is a result of de novo methylation.
Lewis et al.
434 Genome Research
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from

DNA methylation recruits factors that reinforce a repressive
chromatin environment. A methylated-DNA binding activity has
been identified in N. crassa nuclear extracts, consistent with this
possibility (Selker et al. 2002). Interestingly, transcriptional acti-
vation of a silent copy of the bacterial hph gene was observed
following depletion of DNA methylation (Irelan and Selker 1997).
Moreover, loss of H3K9 methylation at a RIP’d allele of the N.
crassa am gene (am
RIP8
) was observed in a dim-2 mutant (Tamaru
et al. 2003). This may indicate that DNA methylation is required
for H3K9 methylation of transcriptionally competent sequences,
but not at sequences that are not typically transcribed. The ex-
tensive RIP of LGVII heterochromatic sequences presumably
inactivated promoter sequences within these domains.
Similarly, HP1 had only a modest influence on the distribu-
tion of H3K9me3. Although we found that HP1 is required to
maintain H3K9 methylation at three loci (i.e., one on LGVII and
two previously identified regions), most heterochromatin do-
mains remain unaffected in an hpo mutant. Specifically, 99.7% of
H3K9me3 on LGVII persists in the hpo mutant. This observation
was surprising because current models implicate HP1 in the
spreading and maintenance of heterochromatin domains in other
organisms. In S. pombe, HP1 is required for spreading of H3K9
methylation across the silent mating type locus and Drosophila
HP1 is required for normal localization of the SU(VAR)3-9 H3K9
KMT (Hall et al. 2002; Schotta et al. 2002). HP1 homologs in
Drosophila and mammals interact with H3K9 KMTs in vivo
(Aagaard et al. 1999; Schotta et al. 2002). Unlike its homologs in S.
pombe, Drosophila, and mammals, DIM-5, the N. crassa H3K9 KMT,
does not have a chromo domain and apparently does not interact
directly with HP1 (Tamaru and Selker 2001; Honda and Selker
2008). Therefore, it is likely that the limited effects of HP1 on
H3K9 methylation are indirect. HP1 may act to recruit histone
deacetylases since treatment of N. crassa with the histone deace-
tylase inhibitor Trichostatin-A results in limited loss of DNA
methylation (Selker 1998).
Epigenetic inheritance reflects the stable maintenance of
a chromatin state through replication cycles independently of the
associated DNA sequence. Reporter genes inserted into the S.
pombe mat locus are stably inherited in either the ‘‘on’’ or ‘‘off’’
state through mitosis and meiosis (Grewal and Klar 1996). In
plants and animals, faithful inheritance of DNA methylation
patterns requires maintenance methylation of hemimethylated
symmetrical sites, such as CpGs, following replication. Indeed,
loss of methylation at CpG sites in A. thaliana met1 mutants is not
completely restored following reintroduction of a wild-type met1
gene, highlighting the importance of existing methylation (Saze
et al. 2003). In contrast, we found no chromosomal regions of N.
crassa that rely on maintenance methylation. This result fits nicely
with our observation that H3K9me3 is normally distributed in
a dim-2 mutant (Fig. 6). Interestingly, we observed complete res-
toration of DNA methylation patterns after depletion and rein-
troduction of the H3K9 methylation machinery, indicating that
the signals for de novo heterochromatin formation lie upstream of
H3K9 methylation. These data indicate that A:T-rich RIP’d DNA
efficiently directs methylation of H3K9 and in turn, directs
methylation of associated cytosines.
Taken together, these results support a model for heterochro-
matin formation in N. crassa that involves efficient targeting of
H3K9 methylation and DNA methylation to A:T-rich relics of RIP.
In contrast to heterochromatin formation in S. pombe and some
DNA methylation in plants, this process appears to be independent
of RNAi and probably relies on DNA binding factors that specifi-
cally recognize A:T-rich DNA. Indeed, the AT-hook analog Dis-
tamycin can inhibit DNA methylation in N. crassa (Tamaru and
Selker 2003). Identification of the factors that direct heterochro-
matin formation at these regions will be critical for developing
a complete model of heterochromatin formation in N. crassa.
Methods
Microarray design
Custom microarrays produced by Agilent Technologies (http://
www.agilent.com) contained 39,386 probes that span LGVII and
2622 control probes corresponding to previously identified
methylated regions. A complete LGVII sequence file was assem-
bled based on the physical map assembled by the Neurospora crassa
genome project (http://www.broad.mit.edu/annotation/genome/
neurospora/Home.html). The relevant contig numbers, contig
orientations, and position on the LGVII sequence are given in
Supplemental Table 3. This assembly includes sequences for the
telomere repeats and the adjacent subtelomeric heterochromatin
for telVIIL, but the telomere repeats and adjacent sequences are
currently not available for telVIIR. Isothermal probes between 45
and 60 bp were designed at intervals of ;100 bp from start to start
as previously described (Thibaud-Nissen et al. 2006; Zilberman
et al. 2007). The microarray contained probes corresponding to
both unique and repeated sequences. It is important to note that
virtually all repeated DNA within the N. crassa genome has been
subjected to RIP. Therefore, repeated sequences typically display
<80% similarity (Galagan and Selker 2004). Of the 39,386 probes
covering LGVII, 38,350 contained at least two or more mis-
matches when compared to the most similar repeated sequence.
Our expectation that the vast majority of our microarray oligo-
nucleotides would not cross hybridize with other regions of the
genome was supported by preliminary results of massive se-
quencing of DNA isolated by MeDIP or ChIP with the antibodies
used in this study (K.M. Smith and M. Freitag, unpubl.).
MeDIP and ChIP
Genomic DNA was isolated for MeDIP as previously described
(Freitag et al. 2004a). MeDIP was performed essentially as de-
scribed except typically 1 mL of antibody specific for 5mC (Dia-
genode) was used to precipitate methylated DNA from 4 mg of total
genomic DNA (Weber et al. 2005). The procedure for ChIP analysis
and antibodies that recognize H3K4me2 (#07-030 Upstate) and
H3K9me3 (a gift from Prim Singh) were previously described
(Tamaru et al. 2003). In addition, ChIP was performed using
a second antibody specific for H3K9me3 (#39161 Active Motif). All
primers used for conventional ChIP analysis are listed in Supple-
mental Table 4.
Sample labeling and microarray hybridization
For MeDIP experiments, ;200 ng of sheared input DNA was la-
beled with Cy3 and 25% of the immunoprecipitated fraction
(from 4 mg input) was labeled with Cy5 using the Bioprime Array
CGH Genomic Labeling system (Invitrogen) as described (Lee et al.
2006). For ChIP-chip experiments, 10 ng of input DNA and 50% of
the immunoprecipitated fraction was amplified using a whole
genome amplification as described (O’Geen et al. 2006). After two
rounds of amplification, 300 ng of input DNA was labeled with
Cy3 and 1 mL of amplified immunoprecipitated DNA was labeled
with Cy5 using the Bioprime Array CGH Genomic Labeling sys-
tem. Microarray hybridizations were performed for ;40 h at 42°C
in hybridization buffer (Lee et al. 2006).
Heterochromatin in N. crassa
Genome Research 435
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from

Data analysis
Microarrays were scanned at 5 mm resolution using a Genepix
4000a scanner (Axon) and pixel intensities were calculated using
Genepix Pro 6.0 (Axon). The data were median normalized and
the log
2
[Cy5/Cy3] ratio was calculated for each spot. Data were
plotted as log
2
values for each spot covering the entire LGVII se-
quence using the Argo genome browser available at http://
www.broad.mit.edu/annotation/argo/. The positions of predicted
open reading frames and repeats were obtained from the Neuros-
pora Genome Project (http://www.broad.mit.edu/annotation/
fungi/neurospora/). To identify statistically significant peaks of
enrichment, we first calculated median values for 500 bp sliding
windows (five spots) to reduce noise. We then utilized the ChIP-
chip data analysis program ChIPOTle v1.11 (Buck et al. 2005) to
call peaks of enrichment. Peaks of 5mC and H3K9me3 were de-
termined for two biological replicate experiments using a window
size of 500 bp and a step size of 100 bp. Replicate experiments for
H3K9me3 were performed with two different antibodies that
recognize H3K9me3 (see above). Only peaks that were detected by
ChIPOTle in both replicate experiments were considered for sub-
sequent analysis. Similarly, we used ChIPOTle to identify peaks of
RIP’d DNA (i.e., regions with high CRI values) using a window size
of 500 bp and a step size of 100 bp. For all analyses, only peaks
with a P-value of 0.05 or better were considered significant.
Strains and culture conditions
N. crassa strains were grown and crossed using standard culture
conditions (http://www.fgsc.net/Neurospora/neurospora.html).
Wild-type strain 74-OR-23-IVA (Selker laboratory strain N150),
dim-2 (strain N1850), hpo (strain N2556), dim-5 hpo-gfp dim-2-
3xflag (strain N3436), hpo-gfp (N3415), and hpo-flag (N3320)
strains were described previously (Kouzminova and Selker 2001;
Freitag et al. 2004a; Honda and Selker 2008). The double Argo-
naute mutant was generously provided by Dr. Yi Liu (UT South-
western). A dim-5 knockout strain (N3074) was created by
replacing the dim-5 gene with a basta-resistance cassette by ho-
mologous recombination as described (Colot et al. 2006). Primers
used to amplify the dim-5 flanking sequence and create a knockout
vector using yeast homologous recombination are listed in Sup-
plemental Table 4. The dim-8 gene was isolated by selecting for
mutants that are defective in methylation (Z.A. Lewis, K.K. Adh-
varyu, S. Honda, and E.U. Selker, unpubl.). Since dim-5 strains are
female sterile, a forced heterokaryon between dim-5 and the
‘‘helper 2’’ strain (FGSC #8745) was used as a female parent in
crosses with dim-8. Helper 2 harbors a deletion of the mating
type locus and therefore does not undergo productive meiosis
(Metzenberg and Sachs 2002).
Acknowledgments
We thank Nicholas Stiffler (UO) for microarray oligo design, Scott
Givan (OSU) for help with oligo design, Reinhard Engels (MIT) for
help with the Argo browser, and Yi Liu (UT Southwestern) for the
double Argonaute mutant. This work was supported by
GM025690-22 to E.U.S. from the National Institutes of Health.
Z.A.L. was supported by fellowships from the American Cancer
Society (PF-04-122-01-GMC) and the Leukemia and Lymphoma
Society (3295-09).
References
Aagaard, L., Laible, G., Selenko, P., Schmid, M., Dorn, R., Schotta, G.,
Kuhfittig, S., Wolf, A., Lebersorger, A., Singh, P.B., et al. 1999. Functional
mammalian homologues of the Drosophila PEV-modifier Su(var)3-9
encode centromere-associated proteins which complex with the
heterochromatin component M31. EMBO J. 18: 1923–1938.
Adhvaryu, K.K., Morris, S.A., Strahl, B.D., and Selker, E.U. 2005.
Methylation of histone H3 lysine 36 is required for normal
development in Neurospora crassa. Eukaryot. Cell 4: 1455–1464.
Amor, D.J., Kalitsis, P., Sumer, H., and Choo, K.H. 2004. Building the
centromere: From foundation proteins to 3D organization. Trends Cell
Biol. 14: 359–368.
Bannister, A.J., Zegerman, P., Partridge, J.F., Miska, E.A., Tho mas, J.O.,
Allshire, R.C., and Kouzarides, T. 2001. Selective recognition of
methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature
410: 120–124.
Bhaumik, S.R., Smith, E., and Shilatifard, A. 2007. Covalent modifications
of histones during development and disease pathogenesis. Nat. Struct.
Mol. Biol. 14: 1008–1016.
Borkovich, K.A., Alex, L.A., Yarden, O., Freitag, M., Turner, G.E., Read, N.D.,
Seiler, S., Bell-Pedersen, D., Paietta, J., Plesofsky, N., et al. 2004. Lessons
from the genome sequence of Neurospora crassa: Tracing the path from
genomic blueprint to multicellular organism. Microbiol. Mol. Biol. Rev.
68: 1–108.
Bostick, M., Kim, J.K., Esteve, P.O., Clark, A., Pradhan, S., and Jacobsen, S.E.
2007. UHRF1 plays a role in maintaining DNA methylation in
mammalian cells. Science 317: 1760–1764.
Buck, M.J., Nobel, A.B., and Lieb, J.D. 2005. ChIPOTle: A user-friendly tool
for the analysis of ChIP-chip data. Genome Biol. 6: R97. doi: 10.1186/gb-
2005-6-11-r97.
Cam, H.P., Sugiyama, T., Chen, E.S., Chen, X., FitzGerald, P.C., and Grewal,
S.I. 2005. Comprehensive analysis of heterochromatin- and RNAi-
mediated epigenetic control of the fission yeast genome. Nat. Gene t. 37:
809–819.
Carrozza, M.J., Li, B., Florens, L., Suganuma, T., Swanson, S.K., Lee, K.K.,
Shia, W.J., Anderson, S., Yates, J., Washburn, M.P., et al. 2005. Histone
H3 methylation by Set2 directs deacetylation of coding regions by
Rpd3S to suppress spurious intragenic transcription. Cell 123: 581–592.
Centola, M. and Carbon, J. 1994. Cloning and characterization of
centromeric DNA from Neurospora crassa. Mol. Cell. Biol. 14: 1510–1519.
Cheutin, T., Gorski, S.A., May, K.M., Singh, P.B., and Misteli, T. 2004. In vivo
dynamics of Swi6 in yeast: Evidence for a stochastic model of
heterochromatin. Mol. Cell. Biol. 24: 3157–3167.
Chicas, A., Forrest, E.C., Sepich, S., Cogoni, C., and Macino, G. 2005. Small
interfering RNAs that trigger posttranscriptional gene silencing are not
required for the histone H3 Lys9 methylation necessary for transgenic
tandem repeat stabilization in Neurospora crassa. Mol. Cell. Biol. 25:
3793–3801.
Cokus, S.J., Feng, S., Zhang, X., Chen, Z., Merriman, B., Haudenschild, C.D.,
Pradhan, S., Nelson, S.F., Pellegrini, M., and Jacobsen, S.E. 2008.
Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA
methylation patterning. Nature 452: 215–219.
Colot, H.V., Park, G., Turner, G.E., Ringelberg, C., Crew, C.M., Litvinkova, L.,
Weiss, R.L., Borkovich, K.A., and Dunlap, J.C. 2006. A high-throughput
gene knockout procedure for Neurospora reveals functions for multiple
transcription factors. Proc. Natl. Acad. Sci. 103: 10352–10357.
Folco, H.D., Pidoux, A.L., Urano, T., and Allshire, R.C. 2008.
Heterochromatin and RNAi are required to establish CENP-A chromatin
at centromeres. Science
319: 94–97.
Foss,
H.M., Roberts, C.J., Claeys, K.M., and Selker, E.U. 1993. Abnormal
chromosome behavior in Neurospora mutants defecti ve in DNA
methylation. Science 262: 1737–1741.
Freitag, M., Hickey, P.C., Khlafallah, T.K., Read, N.D., and Selker, E.U. 2004a.
HP1 is essential for DNA methylation in Neurospora. Mol. Cell 13: 427–
434.
Freitag, M., Lee, D.W., Kothe, G.O., Pratt, R.J., Aramayo, R., and Selker, E.U.
2004b. DNA methylation is independent of RNA interference in
Neurospora. Science 304: 1939. doi: 10.1126/science.1099709.
Fujita, N., Watanabe, S., Ichimura, T., Tsuruzoe, S., Shinkai, Y., Tachibana,
M., Chiba, T., and Nakao, M. 2003. Methyl-CpG binding domain 1
(MBD1) interacts with the Suv39h1-HP1 heterochromatic complex for
DNA methylation-based transcriptional repression. J. Biol. Chem. 278:
24132–24138.
Galagan, J.E. and Selker, E.U. 2004. RIP: The evolutionary cost of genome
defense. Trends Genet. 20: 417–423.
Grewal, S.I. and Jia, S. 2007. Heterochromatin revisited. Nat. Rev. Genet. 8:
35–46.
Grewal, S.I. and Klar, A.J. 1996. Chromosomal inheritance of epigenetic
states in fission yeast during mitosis and meiosis. Cell 86: 95–101.
Hall, I.M., Shankaranarayana, G.D., Noma, K., Ayoub, N., Cohen, A., and
Grewal, S.I. 2002. Establishment and maintenance of a heterochromatin
domain. Science 297: 2232–2237.
Heard, E. and Disteche, C.M. 2006. Dosage compensation in mammals:
Fine-tuning the expression of the X chromosom e. Genes & Dev. 20:
1848–1867.
Lewis et al.
436 Genome Research
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from

Henderson, I.R. and Jacobsen, S.E. 2007. Epigenetic inheritance in plants.
Nature 447: 418–424.
Honda, S. and Selker, E.U. 2008. Direct interaction between DNA
methyltransferase DIM-2 and HP1 is required for DNA methylation in
Neurospora crassa. Mol. Cell. Biol. 28: 6044–6055.
Irelan, J.T. and Selker, E.U. 1997. Cytosine methylation associated with
repeat-induced point mutation causes epigenetic gene silencing in
Neurospora crassa.. Genetics 146: 509–523.
Jackson, J.P., Lindroth, A.M., Cao, X., and Jacobsen, S.E. 2002. Control of
CpNpG DNA methylation by the KRYPTONITE histone H3
methyltransferase. Nature 416: 556–560.
Johnson, L.M., Bostick, M., Zhang, X., Kraft, E., Henderson, I., Callis, J., and
Jacobsen, S.E. 2007. The SRA methyl-cytosine-binding domain links
DNA and histone methylation. Curr. Biol. 17: 379–384.
Kouzminova, E. and Selker, E.U. 2001. dim-2 Encodes a DNA
methyltransferase responsible for all known cytosine methylation in
Neurospora. EMBO J. 20: 4309–4323.
Lachner, M., O ’Carroll, D., Rea, S., Mechtler, K., and Jenuwein, T. 2001.
Methylation of histone H3 lysine 9 creates a binding site for HP1
proteins. Nature 410: 116–120.
Lee, T.I., Johnstone, S.E., and Young, R.A. 2006. Chromatin
immunoprecipitation and microarray-based analysis of protein
location. Nat. Protocols 1: 729–748.
Lehnertz, B., Ueda, Y., Derijck, A.A., Braunschweig, U., Perez-Burgos, L.,
Kubicek, S., Chen, T., Li, E., Jenuwein, T., and Peters, A.H. 2003. Suv39h-
mediated histone H3 lysine 9 methylation directs DNA methylation to
major satellite repeats at pericentric heterochromatin. Curr. Biol. 13:
1192–1200.
Lindroth, A.M., Shultis, D., Jasencakova, Z., Fuchs, J., Johnson, L., Schubert,
D., Patnaik, D., Pradhan, S., Goodrich, J., Schubert, I., et al. 2004. Dual
histone H3 methylation marks at lysines 9 and 27 required for
interaction with CHROMOMETHYLASE3. EMBO J. 23: 4286–4296.
Lister, R., O’Malley, R., Tonti-Filippini, J., Gregory, B., Berry, C., Millar, A.,
and Ecker, J. 2008. Highly integrated single-base resolution maps of the
epigenome in Arabidopsis. Cell 133: 523–536.
Luger, K. 2006. Dynamic nucleosomes. Chromosome Res. 14: 5–16.
Maison, C. and Almouzni, G. 2004. HP1 and the dynamics of
heterochromatin maintenance. Nat. Rev. Mol. Cell Biol. 5: 296–304.
Margolin, B.S., Garrett-Engele, P.W., Stevens, J.N., Fritz, D.Y., Garrett-Engele,
C., Metzenberg, R.L., and Selker, E.U. 1998. A methylated Neurospora 5S
rRNA pseudogene contains a transposable element inactivated by repeat-
induced point mutation. Genetics 149: 1787–1797.
Meissner, A., Mikkelsen, T.S., Gu, H., Wernig, M., Hanna, J., Sivachenko, A.,
Zhang, X., Bernstein, B.E., Nusbaum, C., Jaffe, D.B., et al. 2008.
Genome-scale DNA methylation maps of pluripotent and differentiated
cells. Nature 454: 766–770.
Metzenberg, R.L. and Sachs, M. 2002. Neurospora
heterokaryons involving
a thymidine kinase-positive ‘‘Helper’’: Use in storing poorly viable
strains or crossing strains of limited fertility. Fungal Genet. Newslett. 49:
1–19.
Miao, V.P., Freitag, M., and Selker, E.U. 2000. Short TpA-rich segments of the
z–h region induce DNA methylation in Neurospora crassa. J. Mol. Biol.
300: 249–273.
Miura, A., Yonebayashi, S., Watanabe, K., Toyama, T., Shimada, H., and
Kakutani, T. 2001. Mobilization of transposons by a mutation
abolishing full DNA methylation in Arabidopsis. Nature 411: 212–214.
Nielsen, S.J., Schneider, R., Bauer, U.M., Bannister, A.J., Morrison, A.,
O’Carroll, D., Firestein, R., Cleary, M., Jenuwein, T., Herrera, R.E., et al.
2001. Rb targets histone H3 methylation and HP1 to promoters. Nature
412: 561–565.
O’Geen, H., Nicolet, C.M., Blahnik, K., Green, R., and Farnham, P.J. 2006.
Comparison of sample preparation methods for ChIP-chip assays.
Biotechniques 41: 577–580.
Peng, J.C. and Karpen, G.H. 2008. Epigenetic regulation of heterochromatic
DNA stability. Curr. Opin. Genet. Dev. 18: 204–211.
Perrod, S. and Gasser, S.M. 2003. Long-range silencing and position effects
at telomeres and centromeres: Parallels and differences. Cell. Mol. Life
Sci. 60: 2303–2318.
Reik, W. and Walter, J. 2001. Genomic imprinting: Parental influence on
the genome. Nat. Rev. Genet. 2: 21–32.
Reik, W., Dean, W., and Walter, J. 2001. Epigenetic reprogramming in
mammalian development. Science 293: 1089–1093.
Rountree, M.R. and Selker, E.U. 1997. DNA methylation inhibits elongation
but not initiation of transcription in Neurospora crassa. Genes & Dev. 11:
2383–2395.
Sarraf, S.A. and Stancheva, I. 2004. Methyl-CpG binding protein MBD1
couples histone H3 methylation at lysine 9 by SETDB1 to DNA
replication and chromatin assembly. Mol. Cell 15: 595–605.
Saze, H., Mittelsten Scheid, O., and Paszkowski, J. 2003. Maintenance of
CpG methylation is essential for epigenetic inheritance during plant
gametogenesis. Nat. Genet. 34: 65–69.
Schmiedeberg, L., Weisshart, K., Diekmann, S., Meyer Zu Hoerste, G., and
Hemmerich, P. 2004. High- and low-mobility populations of HP1 in
heterochromatin of mammalian cells. Mol. Biol. Cell 15: 2819–
2833.
Schotta, G., Ebert, A., Krauss, V., Fischer, A., Hoffmann, J., Rea, S., Jenuwein,
T., Dorn, R., and Reuter, G. 2002. Central role of Drosophila SU(VAR)3-9
in histone H3-K9 methylation and heterochromatic gene silencing.
EMBO J. 21: 1121–1131.
Selker, E.U. 1998. Trichostatin A causes selective loss of DNA methylation in
Neurospora. Proc. Natl. Acad. Sci. 95: 9430–9435.
Selker , E.U. and Stevens, J.N. 1987. Signal for DNA methylation associated
with tandem duplication in Neurospora crassa. Mol. Cell. Biol. 7: 1032–
1038.
Selker
, E.U., Fritz, D.Y., and Singer, M.J. 1993. Dense nonsymmetrical DNA
methylation resulting from repeat-induced point muta tion in
Neurospora. Science 262: 1724–1728.
Selker, E.U., Freitag, M., Kothe, G.O., Margolin, B.S., Rountree, M.R., Allis,
C.D., and Tamaru, H. 2002. Induction and maintenance of
nonsymmetrical DNA methylation in Neurospora. Proc. Natl. Acad. Sci.
99 (Suppl. 4): 16485–16490.
Selker, E.U., Tountas, N.A., Cross, S.H., Margolin, B.S., Murphy, J.G., Bird,
A.P., and Freitag, M. 2003. The methylated component of the Neurospora
crassa genome. Nature 422: 893–897.
Sharif, J., Muto, M., Takebayashi, S., Suetake, I., Iwamatsu, A., Endo, T.A.,
Shinga, J., Mizutani-Koseki, Y., Toyoda, T., Okamura, K., et al. 2007. The
SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1
to methylated DNA. Nature 450: 908–912.
Shi, S., Larson, K., Guo, D., Lim, S.J., Dutta, P., Yan, S.J., and Li, W.X. 2008.
Drosophila STAT is required for directly maintaining HP1 localization
and heterochromatin stability. Nat. Cell Biol. 10: 489–496.
Singer, M.J., Marcotte, B.A., and Selker, E.U. 1995. DNA methylation
associated with repeat-induced point mutation in Neurospora crassa.
Mol. Cell. Biol. 15: 5586–5597.
Sullivan, B.A. and Karpen, G.H. 2004. Centrom eric chromatin exhibits
a histone modification pattern that is distinct from both euchromatin
and heterochromatin. Nat. Struct. Mol. Biol. 11: 1076–1083.
Suzuki, M.M. and Bird, A. 2008. DNA methylation landscapes: Provocative
insights from epigenomics. Nat. Rev. Genet. 9: 465–476.
Tamaru, H. and Selker, E.U. 2001. A histone H3 methyltransferase controls
DNA methylation in Neurospora crassa. Nature 414: 277–283.
Tamaru, H. and Selker, E.U. 2003. Synthesis of signals for de novo DNA
methylation in Neurospora crassa.. Mol. Cell. Biol. 23: 2379–2394.
Tamaru, H., Zhang, X., McMillen, D., Singh, P.B., Nakayama, J., Grewal, S.I.,
Allis, C.D., Cheng, X., and Selker, E.U. 2003. Trimethylated lysine 9 of
histone H3 is a mark for DNA methylation in Neurospora crassa. Nat.
Genet. 34: 75–79.
Thibaud-Nissen, F., Wu, H., Richmond, T., Redman, J.C., Johnson, C.,
Green, R., Arias, J., and Town, C.D. 2006. Development of Arabidopsis
whole-genome microarrays and their application to the discovery of
binding sites for the TGA2 transcription factor in salicylic acid-treated
plants. Plant J. 47: 152–162.
Weber, M. and Schubeler, D. 2007. Genomic patterns of DNA methylation:
Targets and function of an epigenetic mark. Curr. Opin. Cell Biol. 19:
273–280.
Weber, M., Davies, J.J., Wittig, D., Oakeley, E.J., Haase, M., La m, W.L., and
Schu
¨
beler, D. 2005. Chromosome-wide and promoter-specific analyses
identify sites of differential DNA methylation in normal and
transformed human cells. Nat. Genet. 37: 853–862.
Weber, M., Hellmann, I., Stadler, M.B., Ramos, L., Pa
¨
a
¨
bo,
S., Rebhan, M.,
and Schu
¨
beler, D. 2007. Distribution, silencing potential and
evolutionary impact of promoter DNA methylation in the human
genome. Nat. Genet. 39: 457–466.
Wu, C., Smith, K., Li, W., Hood, H., Sachs, M., Staben, C., Selker, E., and
Farman, M. 2008. Characterization of chromosome ends in the
filamentous fungus Neurospora crassa. Genetics (in press). doi: 10.1534/
genetics.107.084392.
Zhang, X., Yazaki, J., Sundaresan, A., Cokus, S., Chan, S.W., Chen, H.,
Henderson, I.R., Shinn, P., Pellegrini, M., Jacobsen, S.E., et al. 2006.
Genome-wide high-resolution mapping and functional analysis of DNA
methylation in Arabido psis. Cell 126: 1189–1201.
Zhang, K., Mosch, K., Fischle, W., and Grewal, S.I. 2008. Roles of the Clr4
methyltransferase complex in nucleation, spreading and maintenance
of heterochromatin. Nat. Struct. Mol. Biol. 15: 381–388.
Zilberman, D., Gehring, M., Tran, R.K., Ballinger, T., and Henikoff, S. 2007.
Genome-wide analysis of Arabidopsis thaliana DNA methylation
uncovers an interdependence between methylation and transcription.
Nat. Genet. 39: 61–69.
Received September 8, 2008; accepted in revised form December 8, 2008.
Heterochromatin in N. crassa
Genome Research 437
www.genome.org
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from

10.1101/gr.086231.108Access the most recent version at doi:
2009 19: 427-437 originally published online December 17, 2008Genome Res.
Zachary A. Lewis, Shinji Honda, Tamir K. Khlafallah, et al.
Neurospora crassaformation in
Relics of repeat-induced point mutation direct heterochromatin
Material
Supplemental
http://genome.cshlp.org/content/suppl/2009/02/06/gr.086231.108.DC1
References
http://genome.cshlp.org/content/19/3/427.full.html#ref-list-1
This article cites 75 articles, 30 of which can be accessed free at:
License
Service
Email Alerting
click here.top right corner of the article or
Receive free email alerts when new articles cite this article - sign up in the box at the
https://genome.cshlp.org/subscriptions
go to: Genome Research To subscribe to
Copyright © 2009 by Cold Spring Harbor Laboratory Press
Cold Spring Harbor Laboratory Press on September 17, 2024 - Published by genome.cshlp.orgDownloaded from
