
The H3K4me1 histone mark recruits DNA repair to functionally
constrained genomic regions in plants
Daniela Quiroz
1,2
, Diego Lopez-Mateos
4,5
, Kehan Zhao
1,3
, Alice Pierce
1,3
, Lissandro Ortega
1
, Alissza Ali
1
, Pablo
Carbonell-Bejerano
5
, Vladimir Yarov-Yarovoy
4,5
, J. Grey Monroe
1,2,3,#
1
Department of Plant Sciences, University of California Davis, Davis, CA, USA 95616
2
Integrative Genetics and Genomics, University of California Davis, Davis, CA, USA 95616
3
Plant Biology Graduate Group, University of California Davis, Davis, CA, USA 95616
4
Department of Physiology and Membrane Biology, University of California Davis, Davis, CA, USA 95616
5
Biophysics Graduate Group, University of California Davis, Davis, California
6
Institute for Grape and Wine Sciences (ICVV, CSIC-CAR-UR), 26007 Logroño, La Rioja, Spain
#
Correspondence: gmonr[email protected]
Abstract
Mutation is the ultimate source of genetic variation. Mutation rate variability has been observed within plant
genomes, but the underlying mechanisms have been unclear. We previously found that mutations occur less often in
functionally constrained regions of the genome in Arabidopsis thaliana and that this mutation rate reduction is
predicted by H3K4me1, a histone modi
fication found in the gene bodies of actively expressed and evolutionarily
conserved genes in plants. We reanalyzed de novo germline single base substitutions in fast neutron irradiated
mutation accumulation lines in Kitaake rice (Oryza sativa) and found the same reduction in mutations associated with
H3K4me1, gene bodies, and constrained genes as in A. thaliana, suggesting conserved mechanisms for mutation
reduction in plants. Here, we characterize a model of targeted DNA repair to explain these observations; PDS5C and
MSH6 DNA repair-related proteins target H3K4me1 through their Tudor domains, resulting in nearby DNA
experiencing elevated repair. Experimental data and in-silico modeling support the high a
ffinity of the Tudor domain
for H3K4me1 in both proteins, and that this a
ffinity is conserved between plant species. ChIP-seq data from PDS5C
con
firms its localization to conserved and low mutation rate genome regions. Somatic and germline mutations
observed by deep sequencing of wild-type and MSH6 knockout lines con
firm that MSH6 preferentially repairs gene
bodies and H3K4me1-enriched regions. These fi
ndings inspire further research to characterize the origins of
mechanisms of targeted DNA repair in eukaryotes and their consequences on tuning the evolutionary trajectories of
genomes.
Introduction
Mutations occur when DNA damage or replication
error goes unrepaired. Mechanisms that localize DNA repair
proteins to certain genome regions, such as by binding
certain histone modi
fications, reduce local mutation rates.
Interactions between DNA repair and histone modi
fications
are predicted to evolve if they promote repair in regions
prone to deleterious mutations, such as coding regions of
essential genes (Lynch, 2010; Martincorena and Luscombe,
2013; Lynch et al., 2016).
Associations between histone modi
fications and
mutation rates have been observed across diverse
organisms (Habig et al., 2021; de la Peña et al., 2022; Yang
et al., 2021; Yan et al., 2021; Monroe et al., 2022; Makova
and Hardison, 2015; Schuster-Böckler and Lehner, 2012).
The localization of DNA repair proteins responsible for such
mutation biases has been well-established in humans
(Supek and Lehner, 2015, 2017, 2019; Foster et al., 2015;
Katju et al., 2022). In vertebrates, H3K36me3 is targeted by
PWWP domains in proteins contributing to
homology-directed and mismatch repair, with H3K36me3
marking the gene bodies and exons of active genes (Li et
al., 2013; Huang et al., 2018; Fang et al., 2021; Aymard et
al., 2014; Sun et al., 2020). As predicted, reduced mutation
rates in active genes and gene bodies have been observed
in humans and other animals (Moore et al., 2021; Akdemir et
al., 2020; Li et al., 2021; Katju et al., 2022; Supek and
Lehner, 2017).
1The author(s) responsible for distribution of materials integral to the findings presented in this article in accordance with the policy described
in the Instructions for Authors (https://academic.oup.com/plcell/pages/General-Instructions) is (are): Grey Monroe ([email protected]).
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
Reduced mutation rates in gene bodies and active
genes have also been observed in some algae and land
plants, but the mechanism has been unclear (Bel
field et al.,
2021; Lu et al., 2021; Zhu et al., 2021; Monroe et al., 2022;
Bel
field et al., 2018; López-Cortegano et al., 2021; Yan et
al., 2021; Krasovec et al., 2017). Knockout lines of MSH2,
the mismatch repair protein that dimerizes with MSH6 to
form MutSα (Adé et al., 1999; Kolodner, 1996), indicate that
mismatch repair can preferentially target gene bodies in
plants. Yet, a speci
fic mechanism of such targeting is
unresolved (Bel
field et al., 2018).
In plants, H3K4me1 marks gene bodies of active
genes. Recent work has demonstrated that H3K4me1
enrichment is mediated by a combination of
transcription-coupled (ATXR7) and epigenome-guided
(ATX1, ATX2) methyltransferases (Oya et al., 2021). Once
established, H3K4me1 reading can occur by proteins
containing histone-reader domains such as “Royal family”
Tudor domains, which bind methylated lysine residues on
H3 histone tails (Kim et al., 2006; Lu and Wang, 2013;
Maurer-Stroh et al., 2003).
Recently, the Tudor domain of PDS5C was shown
to speci
fically bind H3K4me1 in A. thaliana (Niu et al., 2021).
This gene is also a cohesion cofactor that facilitates
homology-directed repair (Pradillo et al., 2015; Phipps and
Dubrana, 2022; Morales et al., 2020). Recent studies of
CRISPR-mediated mutation e
fficiency show that H3K4me1
is associated with lower mutation e
fficacy (
R = -0.64),
supporting more e
fficient repair (Weiss et al., 2022; Schep et
al., 2021; Zhu et al., 2021). These fi
ndings are consistent
with analyses of mutation accumulation lines in A. thaliana,
which fi
nd H3K4me1 associated with lower mutation rates
(Monroe et al., 2022). Still, the extent and consequences of
targeted DNA repair remain contentious (Liu and Zhang,
2022).
Here, we reanalyzed de novo mutations from
whole-genome-sequenced fast neutron mutation
accumulation lines in Kitaake Rice (O. sativa) (Li et al., 2017)
showing similar patterns of mutation reduction and
H3K4me1 association as A. thaliana. We then characterize a
mechanistic model to explain these observations, involving
PDS5C and MSH6 proteins targeting H3K4me1 via Tudor
domains.
Results and Discussion
Mutations in O. sativa FN-irradiated lines re
flect no
selection on SBS mutations
Mutagenesis has been used extensively in the
generation and study of plant mutation. With single base
substitutions (SBS) in fast-neutron mutation accumulation
lines largely re
flecting native mutational processes (Wyant et
al., 2022; Li et al., 2017), the distribution of these de novo
mutations could provide insights into mechanisms
underlying intragenomic heterogeneity in mutation rate.
We fi
rst reanalyzed de novo mutations in a
population of 1,504 O. sativa lines that accumulated
mutations upon fast neutron radiation. These data were
previously described, and single base-pair substitutions
(SBS) were validated with a >99% true positive rate (Li et al.,
2017). In total, these data included 43,483 SBS, comprising
a combination of indistinguishable fast neutron-related and
“spontaneous” mutations (Fig. 1). We restricted our
analyses to SBS mutations to preclude selection that may
have occurred in the generation of these lines on insertions,
deletions, and structural variants (i.e., whole gene deletions).
To quantitatively evaluate whether selection on SBS
mutations occurred in these lines, we examined
non-synonymous and synonymous mutation rates. The ratio
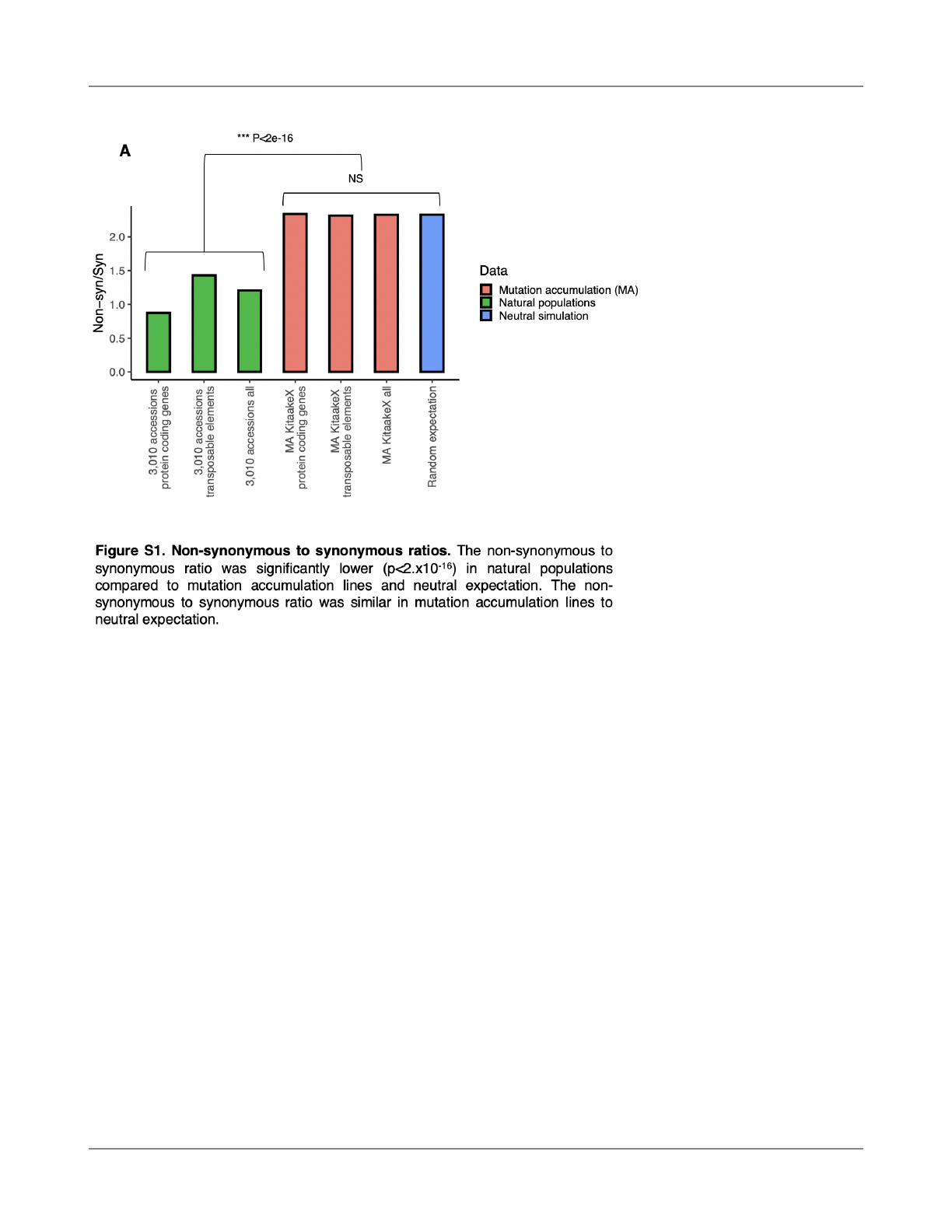
of non-synonymous to synonymous mutations in mutation
accumulation lines was 2.33 (N/S=5,370/2,155), a 190%
increase over this ratio (Pn/Ps = 1.21) observed in
polymorphisms of 3,010 sequenced O. sativa accessions
(Wang et al., 2018) (X
2
= 670.63, p<2x10
-16
). The ratio of
non-synonymous to synonymous de novo mutations was
not higher in transposable elements (TE) (N/S=2.31) than in
non-TE protein-coding genes (N/S=2.34) (X
2
= 0.035, p =
0.85) nor was it less in coding genes than neutral
expectations (N/S=2.33) based on mutation spectra and
nucleotide composition of coding regions in the O. sativa
genome (X
2
= 0.029, df = 1, p = 0.86) (Fig. S1, methods). To
con
firm that this non-significance was not simply due to a
lack of power, we tested how many non-synonymous
mutations would have to be “missing” because of selection
to explain the 29% reduction in mutation rate of coding
genes relative to non-genic (intergenic, TE) regions we
observed in downstream analyses. Given that coding
regions constitute only 37% of gene bodies (the rest being
intronic and untranslated exonic regions), we fi
nd that 34%
of non-synonymous mutations would have to have been
missing due to selection, which would be readily detected
by a signi
ficant deviation from the null expectation (X
2
=
224.4, df = 1, p < 2e-16). In conclusion, we detect no
evidence of selection on SBS mutations, providing an
opportunity to study de novo mutation rate heterogeneity
before selection.
We then compared SBS spectra in trinucleotide
contexts from these lines with de novo germline mutations
in A. thaliana (Weng et al., 2019; Monroe et al., 2022) and
found that they are signi
ficantly correlated (Fig. 1A, r=0.8,
p<2x10
-16
). We cannot know how much of the residual
2
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
di
fference in SBS spectra is due to the effects of fast
neutron mutagenesis versus natural di
fferences between O.
sativa and A. thaliana, as variations in the spectra of SBS
have been reported between related species and even
di
fferent genotypes of the same species (Jiang et al., 2021;
Sasani et al., 2022; Cagan et al., 2022). Future work will
bene
fit from the evaluation of de novo mutations arising in
di
fferent species under diverse conditions to understand the
environmental and genetic controls of SBS mutational
spectra in plants.
H3K4me1 marks low mutation rate regions in O. sativa,
including gene bodies, and transcriptionally active and
evolutionarily conserved genes
To test whether the genome-wide distribution of
mutations in O. sativa is associated with histone
modi
fications, we used data from the riceENCODE
epigenomic database, which includes H3K4me1, H3K9me1,
H3K4me3, H3K36me3, H3K9me2, H3K27me3, H3K27ac,
H3K4ac, H3K12ac, H3K9ac, and RNA polymerase II (PII)
measured by chromatin immunoprecipitation sequencing
(ChIP-seq) (Xie et al., 2021). We then tested whether
mutation probabilities in 100bp windows in genic regions
(the probability of observing a mutation) were predicted by
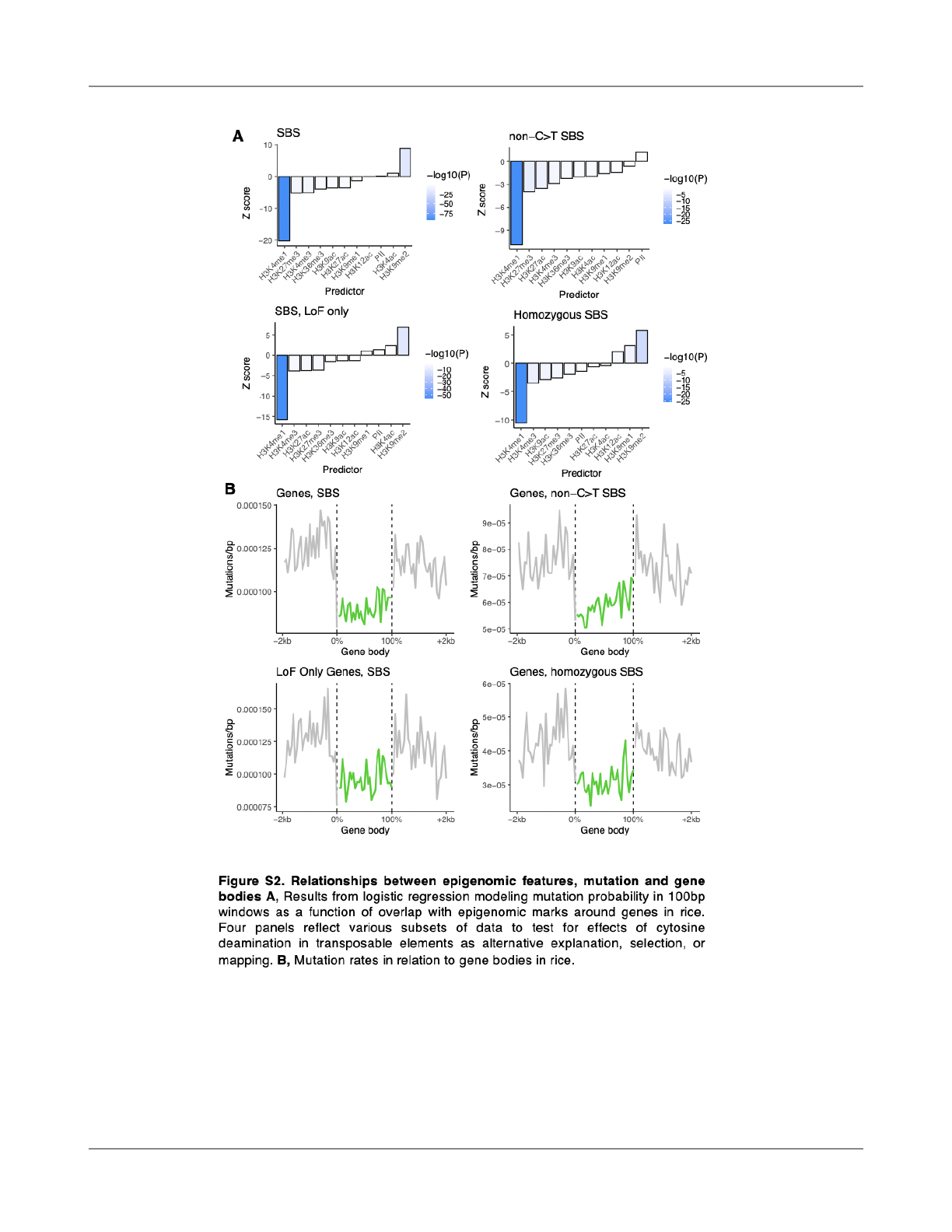
epigenomic features and found a signi
ficant reduction in
mutation probabilities in windows that overlapped with
H3K4me1 peaks (Fig. 1B). These data are consistent with
observations in other plant species where lower mutation
rates have been observed in regions marked by H3K4me1
(Monroe et al., 2022; Weiss et al., 2022). To further con
firm
that these patterns were not due to selection, we also
restricted our analyses to only those genes in which
loss-of-function mutations were found in the population (Li
et al., 2017) and observed similar results (Fig. S2). We
considered that mutation rate heterogeneity was possibly
caused by GC>AT mutations in transposable elements with
3
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
elevated cytosine methylation in non-genic sequences
rather than histone-mediated mutation reduction. Therefore,
we restricted our analyses to exclude all such GC>AT
mutations and observed similar results.
H3K4me1-associated hypomutation was also the same
when analyses were restricted to only homozygous
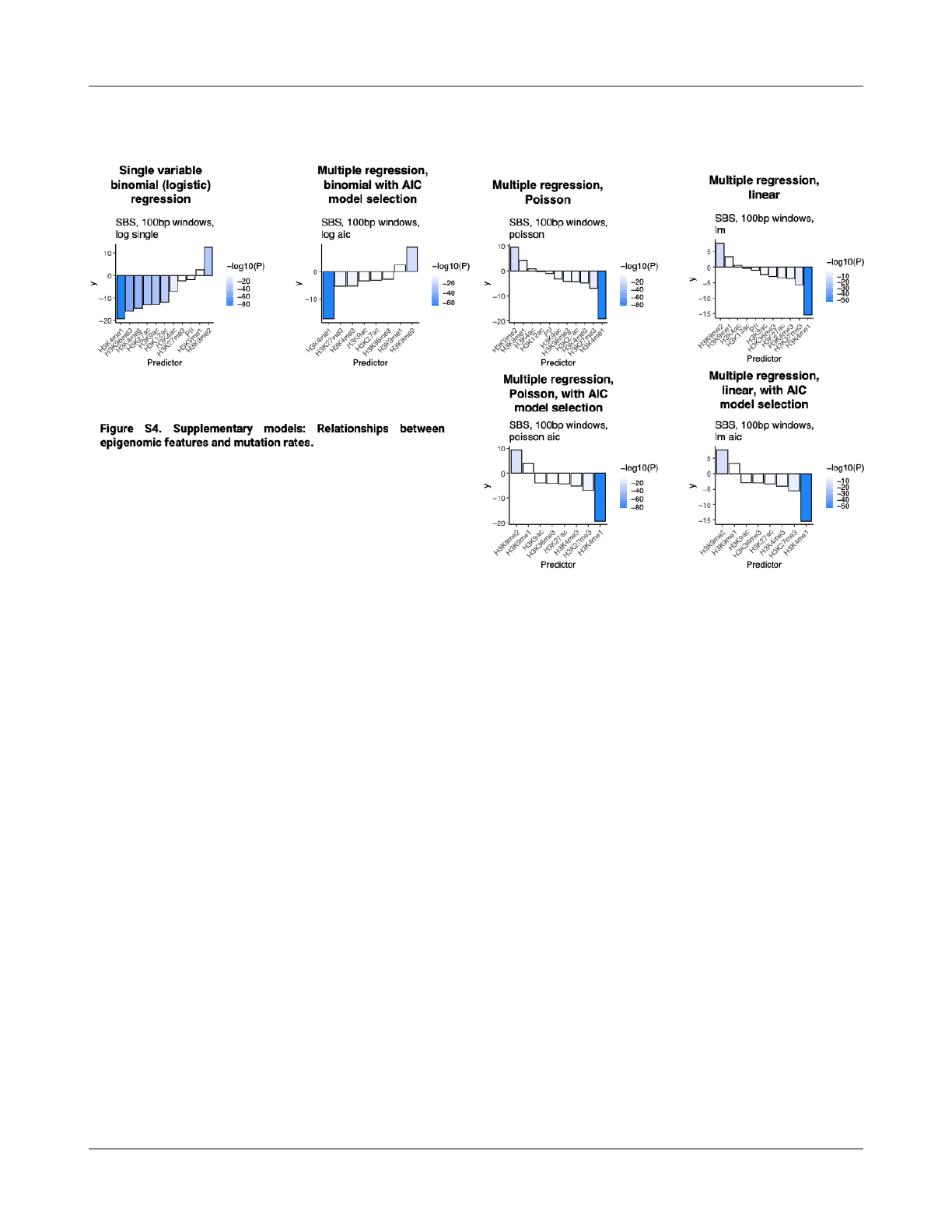
mutations (Fig. S2). We further repeated analyses with other
regression approaches: single predictor binomial regression,
multiple linear regression, Poisson regression, and model
selection based on AIC. In all cases, H3K4me1 was found to
have the most signi
ficant association with reduced mutation
rates (Fig. S4)
We calculated mutation rates in genes and their
neighboring sequences and observed a signi
ficant reduction
in mutation rates in gene bodies (Fig. 1C), consistent with
lower mutation rates in gene bodies of other plants.
Mutation rates were lower both in and around H3K4me1
peaks, which could indicate the action of local recruitment
and targeting of DNA repair to H3K4me1 marked sequences
including gene bodies (Fig. 1D). That mutation rates were
also lower in sequences immediately neighboring H3K4me1
peaks could indicate a spatially distributed e
ffect on
mutation around H3K4me1, or the e
ffect of conservative
peak calling (Fig. 1D). Only 8.9% of H3K4me1 peaks were
found outside of non-TE protein-coding genes.
Nevertheless, we could use these instances of non-genic
H3K4me1 to test whether the reduction in mutation rates in
H3K4me1 peaks was due simply to selection against coding
region mutations a
ffecting results. When considering all
H3K4me1 peaks, we observed a 20.1% reduction in
mutation rates compared to regions within 2kb outside of
peaks (X
2
= 124.38, p < 2.2e-16 -). For non-genic H3K4me1
peaks, we observed the same reduction: -20.2% (X
2
= 9.88,
p = 0.00167). Together, these results suggested a role of
H3K4me1 in localized hypomutation, which could explain
reduced gene body mutation rates observed since gene
bodies are enriched for H3K4me1 (Fig. 1E,F).
We then compared mutation rates between
di
fferent classes of genes, which proved consistent with the
expected e
ffects of increased DNA repair in functionally
constrained genes caused by H3K4me1-localized repair.
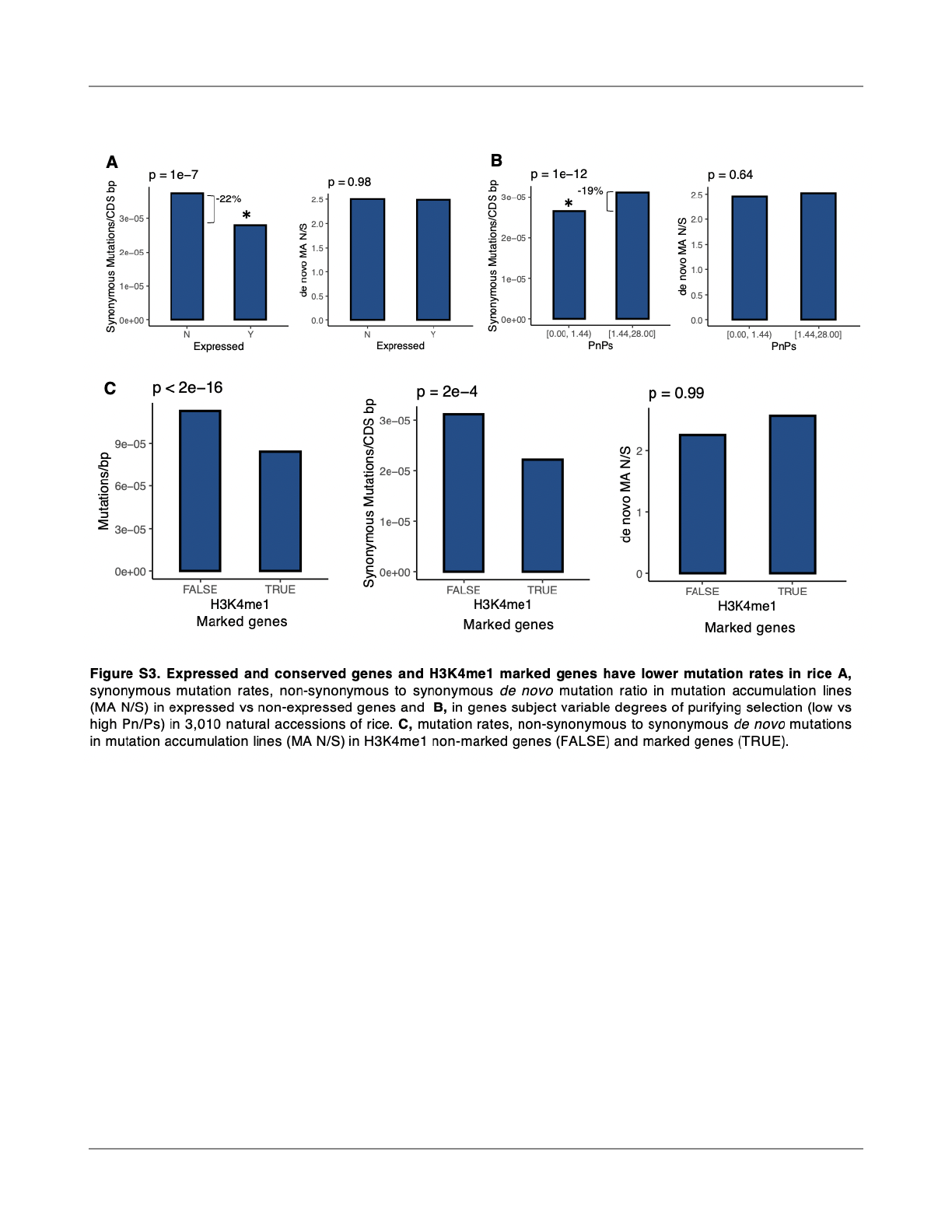
Mutation rates were signi
ficantly lower in genes that
overlapped with H3K4me1 peaks, including when
considering only synonymous mutations (Fig S3). H3K4me1
peaks were enriched in genes annotated as expressed
compared with those not expressed (Kawahara et al., 2013)
(X
2
= 2550961, p < 2x10
-16
). And, as predicted by their
enrichment for H3K4me1, mutation rates were 22% lower in
expressed genes (X
2
= 63.7, p = 1x10
-15
)(Fig. 1G). This result
was con
firmed in an analysis restricted to synonymous
mutations in coding regions only, rejecting the hypothesis
that these results are due to selection (Fig. S3). Indeed, the
ratio of non-synonymous to synonymous de novo mutations
in the data was not di
fferent between expressed and
non-expressed genes (X
2
= 0.0007, p = 0.98) (Fig. S3).
Comparing genes that exhibit di
fferent degrees of selection
in 3,010 natural accessions of O. sativa (Wang et al., 2018),
those under elevated purifying selection with low Pn/Ps
(non-synonymous/synonymous polymorphisms), were
enriched for H3K4me1 peaks (X
2
= 8045711, p < 2x10
-16
)
and experienced 19% lower mutation rates (X
2
= 188.5, p <
2x10
-16
) (Fig. 1H). These genes did not have a lower ratio of
non-synonymous to synonymous in de novo mutations (X
2
=
0.22, p=0.63) (Fig S3) and the reduction of mutation rates in
conserved genes was con
firmed by analysis of synonymous
mutations only (Fig S3). We, therefore, fi
nd that mutation
rates are signi
ficantly lower in functionally constrained
genes in O. sativa, which cannot be explained by selection
on non-synonymous mutations, consistent with what has
been previously shown in A. thaliana (Monroe et al., 2022).
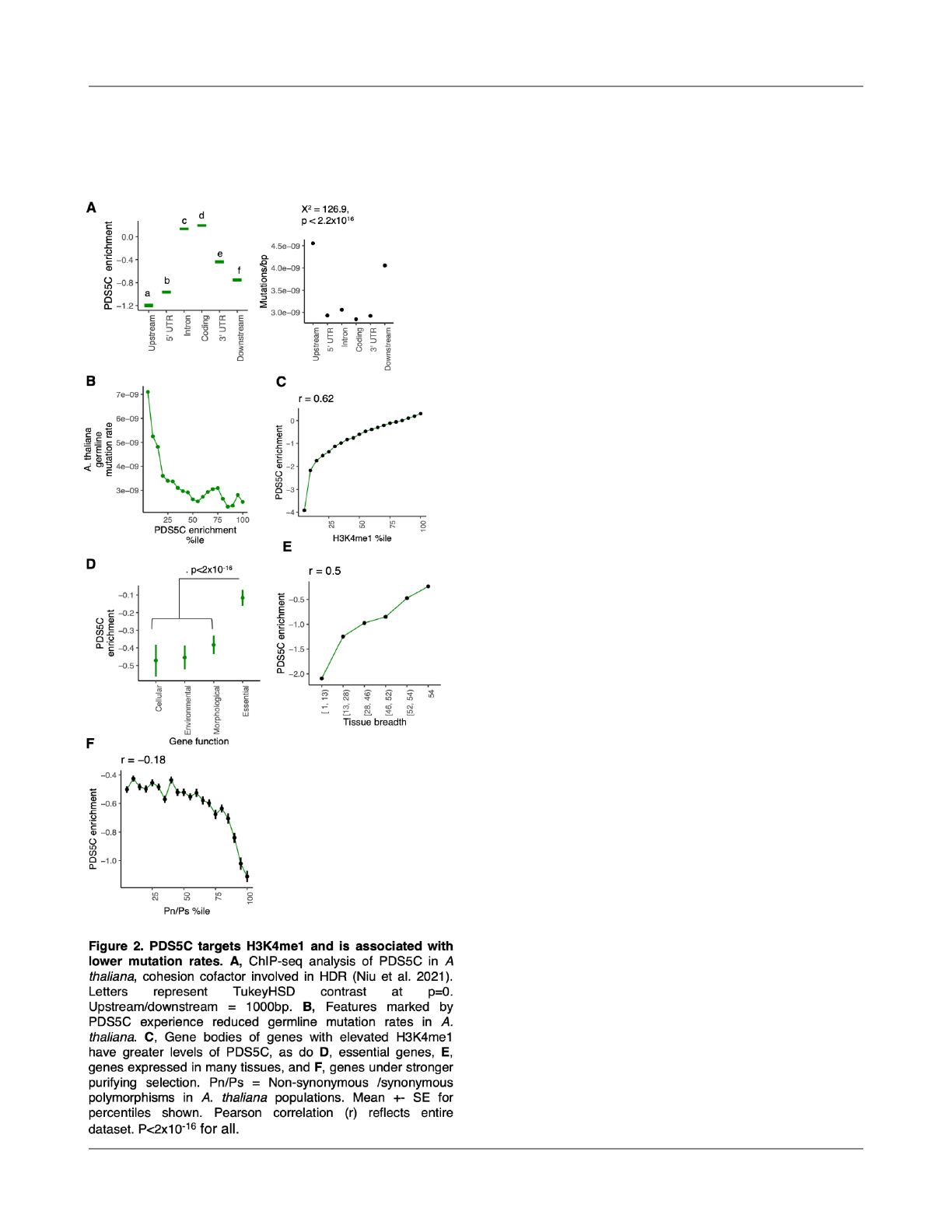
PDS5C targets H3K4me1 and is associated with lower
mutation rates
These fi
ndings in O. sativa are consistent with
reports of reduced mutation rates in gene bodies of
expressed and constrained genes in A. thaliana and other
species (Krasovec et al., 2017; Moore et al., 2021; Monroe
et al., 2022). While in humans, this is known to be mediated
by H3K36me3 targeting by DNA repair genes, our results
suggest that H3K4me1 may be a target of DNA repair in
plants. To examine this further, we considered genes with
known H3K4me1 targeting. PDS5C, a gene belonging to a
family of cohesion cofactors that facilitate
homology-directed repair (HDR), contains a Tudor domain
that was recently discovered to speci
fically bind H3K4me1
(Niu et al., 2021). Analyses of ChIP-seq data of PDS5C-Flag
from A. thaliana show PDS5C is targeted to gene bodies
(which are enriched for H3K4me1 in both O. sativa and A.
thaliana) (Fig. 2A). We also fi
nd that PDS5C is increased in
regions of lower germline mutation rates in A. thaliana,
consistent with recruitment and its function in facilitating
DNA repair (Fig. 2B).
Evolutionary models predict that histone-mediated
repair mechanisms should evolve if they facilitate lower
mutation rates in sequences under purifying selection
(Lynch et al., 2016; Martincorena and Luscombe, 2013). As
predicted by this theory, we fi
nd that PDS5C targeting
(ChIP-seq) is enriched in coding sequences, essential genes
(determined by experiments of knockout lines (Lloyd and
Meinke, 2012; Lloyd et al., 2015)), genes constitutively
expressed (detected in 100% of tissues sampled) (Mergner
et al., 2020), and genes under stronger purifying selection in
4
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
natural populations of A. thaliana (lower Pn/Ps) (1001
Genomes Consortium, 2016) (Fig. 2C-F).
Visualizing the A. thaliana PDS5C protein full-length
model generated by Alphafold (Jumper et al., 2021) reveals
that the PDS5C active domain is separated from the Tudor
domain by unstructured, and potentially fl
exible segments
(Fig. 3A), suggesting that the Tudor domain operates as an
anchor, localizing PDS5C to H3K4me1 and gene bodies of
active genes. PDS5C is a cohesion cofactor linked to
multiple DNA repair pathways. In its role in cohesion
between sister chromatids, it has been reported to promote
HDR (Pradillo et al., 2015). This is consistent with its known
interaction with repair proteins along with the direct
contribution of cohesion and PDS orthologs to the HDR
pathway (Morales et al., 2020; Phipps and Dubrana, 2022;
Hill et al., 2016; Ren et al., 2005; Bolaños-Villegas et al.,
2013; Schubert et al., 2009). The observation that mutation
rates are reduced at H3K4me1 peak regions (Fig. 1)
supports the hypothesis that Tudor domain-mediated
targeting in PDS5C, its orthologs (PDS5A, B, D, and E), or
other repair-related proteins contribute to targeted
hypomutation in the functionally important regions of the
genome. Still, additional experiments are needed to quantify
the precise local e
ffect of PDS5C on mutation rate. We
compared the PDS5C Tudor domain sequence between A.
thaliana and O. sativa and found that the critical amino acids
constituting the aromatic cage, where H3K4me1 binding
speci
ficity is determined, are conserved (Fig. 3C),
suggesting a potential role of PDS5C in the mutation biases
observed here in O. sativa (Fig. 1).
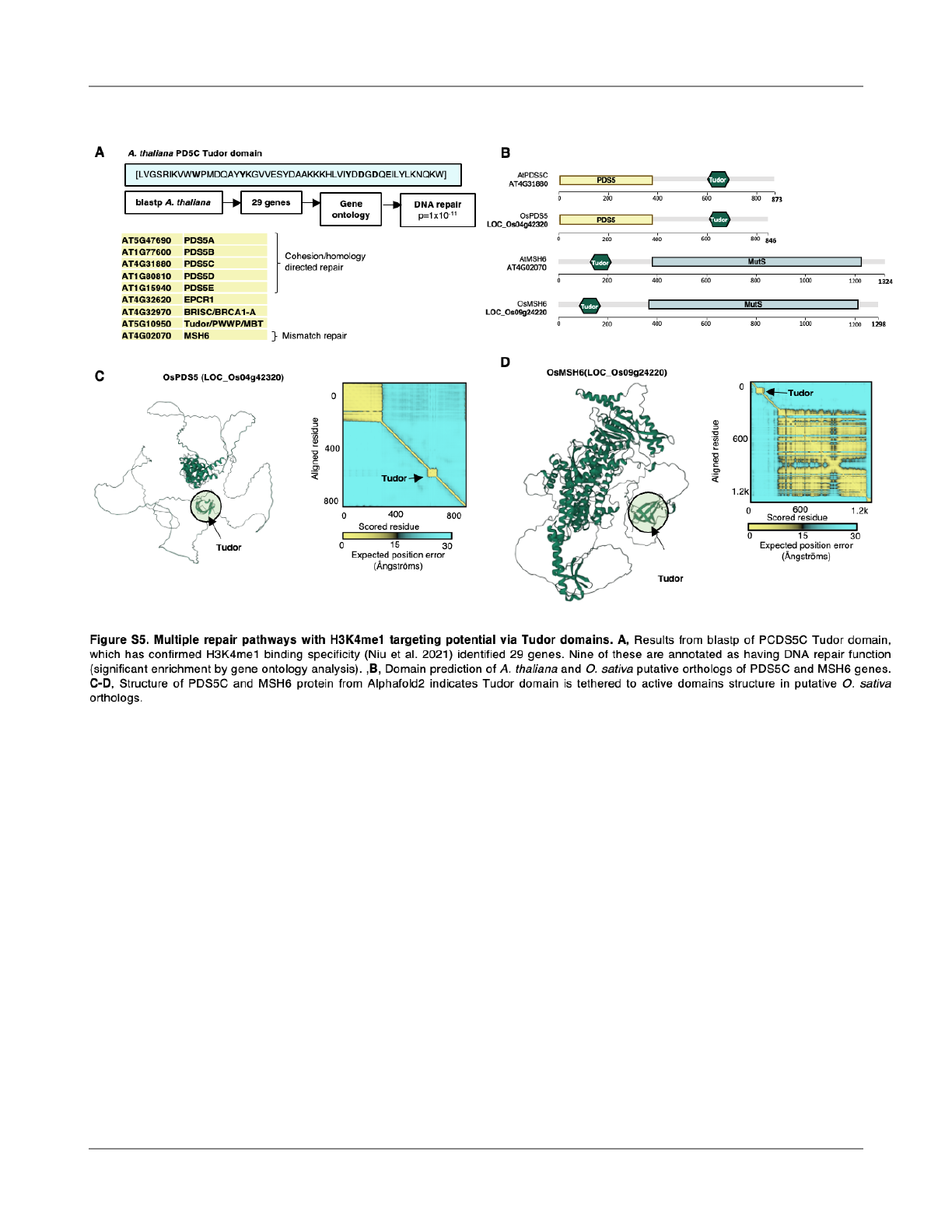
Multiple DNA repair mechanisms with Tudor domains
could be in
fluencing mutation biases
The discovery of the PDS5C Tudor domain as an
H3K4me1 targeting domain (Niu et al., 2021) provides an
opportunity to identify other proteins with potential for
H3K4me1-mediated gene body recruitment. We used blastp
to search the A. thaliana proteome for other proteins
containing Tudor domains similar to that of PDS5C. An
analysis of gene ontologies indicated that this gene set is
highly enriched for genes with DNA repair functions (9/29
genes, p=1x10
-11
)(Fig. S5A, Table S1). Five of these were
PDS5C homologs. We also found that MSH6, a DNA
mismatch repair protein, contains a Tudor domain similar to
that of PDS5C, which was an obvious candidate for further
consideration. That the MSH6 homologous protein in
vertebrates instead contains a PWWP that targets gene
bodies via H3K36me3 binding suggests a remarkable
example of convergent evolution between plants and
vertebrates (Li et al., 2013).
Structural modeling prediction of MSH6 structure
using AlphaFold (Jumper et al., 2021) indicates that its Tudor
domain may, like that in PDS5C, function as an anchor,
tethering it to H3K4me1 leading to local increases in DNA
repair (Fig. 3A-B, Fig. S5). Sequence comparison suggests
5
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
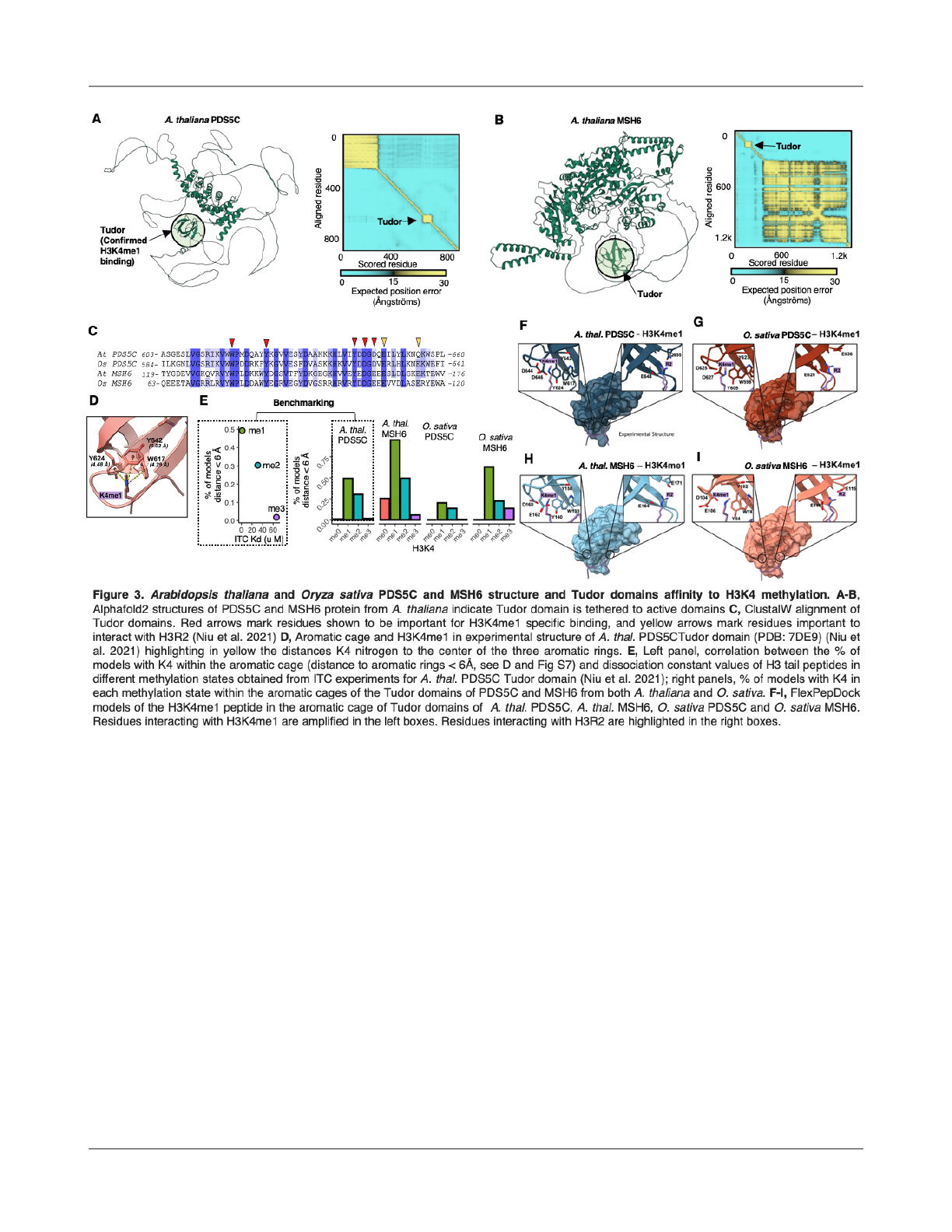
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
that O. sativa MSH6, A. thaliana MSH6, and O. sativa
PDS5C Tudor domains may present a similar binding
preference for H3K4me1 as A. thaliana PDS5C Tudor
domain since key residues forming monomethylated lysine
binding site are conserved between all homologous
domains (Fig. 3C). To test this hypothesis, we modeled A.
thaliana MSH6 and O. sativa MSH6 and PDS5C Tudor
domains with AlphaFold and compared them with the
experimental structure of A. thaliana PDS5C Tudor domain
(PDB:7DE9) (Niu et al., 2021). Superimposition of the three
modeled Tudor domains onto the PDS5C Tudor domain
showed remarkably similar structures with backbone root
mean square deviation (RMSD) values below 1 Å (Fig. S6).
Subsequently, we modi
fied the K4me1 from the H3
tail peptide bound to the PDS5C Tudor domain in ChimeraX
(Goddard et al., 2018) to obtain H3K4, H3K4me2, and
H3K4me3. Using Rosetta FlexPepDock (Raveh et al., 2010),
we modeled the docking of the di
fferent methylation states
of H3K4 to the Tudor domains of PDS5C and MSH6 from A.
thaliana and O. sativa.
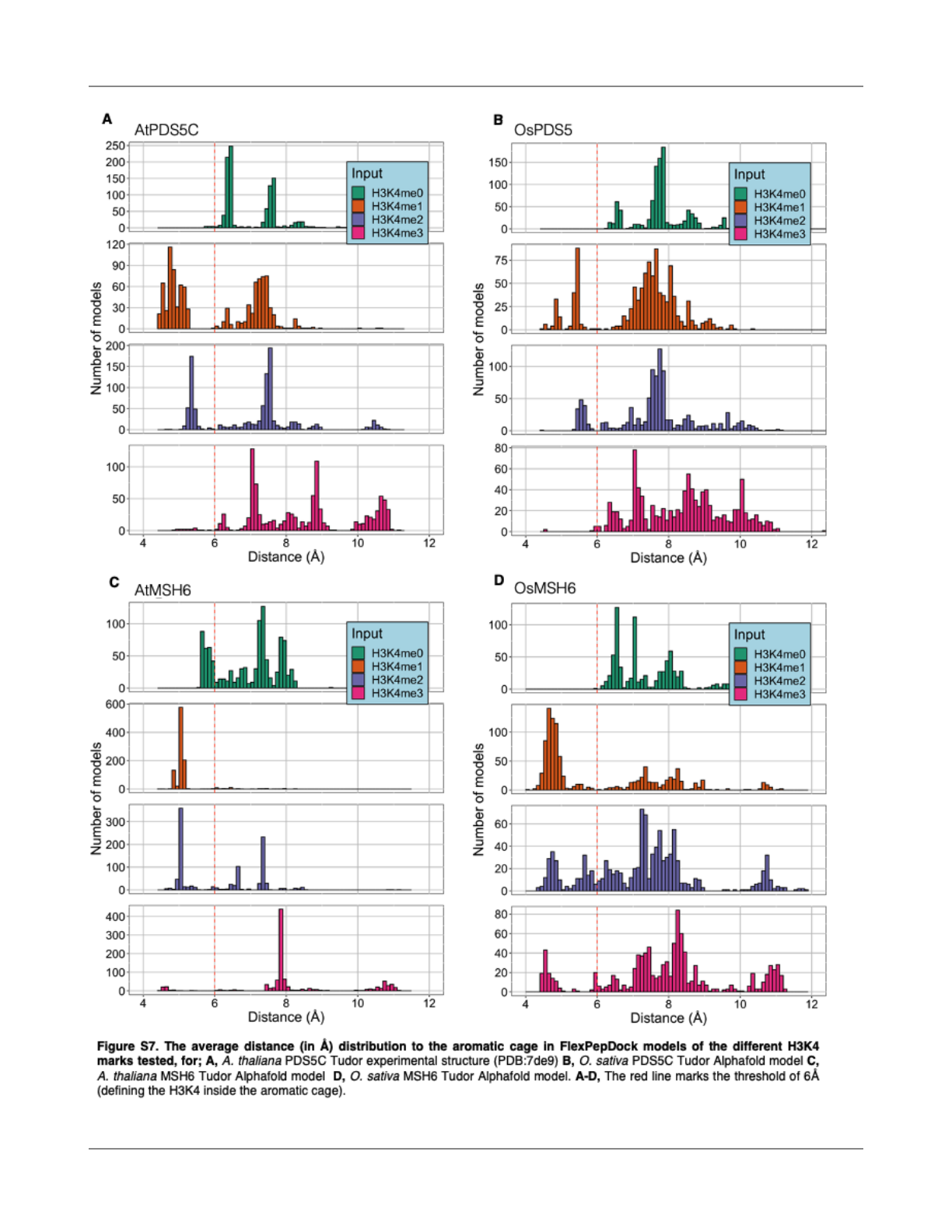
We analyzed the geometry of the binding site in the
top 10% of models based on Rosetta total score, by
measuring the distances of H3K4 nitrogen to the center of
the three aromatic rings forming the aromatic cage in the
Tudor domains (Fig 3D). The experimental structure of the
PDS5C Tudor domain bound to H3K4me1 (PDB:7DE9) (Niu
et al., 2021) provided a reference to analyze the geometry of
the aromatic cage. In this structure, the average distance
between H3K4 nitrogen and the aromatic rings is 4.6 Å (Fig.
3D). We calculated this average distance in the in silico
models generated by FlexPepDock and analyzed its
distributions per case (Fig. S7).
We found that by setting a distance threshold of
6Å, we were able to accurately calculate the proportion of
generated models with the H3K4 inside the aromatic cage.
For the A. thaliana PDS5C Tudor, the proportion of models
with H3K4 inside the aromatic cage was a near-perfect
6
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
correlation (r=0.999) with the dissociation constant values
obtained experimentally by ITC (Figure 3E). Therefore, we
reasoned that we could apply this approach to predict the
binding preference of the Tudor domains of A. thaliana
MSH6 and O. sativa MSH6 and PDS5C to the di
fferent
epigenetic marks. For all three cases, we observed that
H3K4me1 was the mark sampled more often inside the
aromatic cage (Fig. 3E), followed by H3K4me2, and
H3K4me3, aligning with the experimental results for the
Tudor domain of A. thaliana PDS5C. Interestingly, lack of
methylation resulted in almost no sampling of geometries
within the aromatic cage, which aligns with ITC experimental
results that show no detectable binding of H3 tail peptide by
the PDS5C Tudor domain when K4 was not methylated (Niu
et al., 2021). These results suggest that MSH6 and PDS5C
Tudor domains bind to H3K4 with more a
ffinity when it is in
the monomethylated state (H3K4me1). While binding to
H3K4me2 and me3 is possible, our results suggest a larger
energetic barrier to access the aromatic cage for these two
states and lower a
ffinity. However, binding experiments
must be conducted to obtain dissociation constant values,
thus determining the exact selectivity for H3K4me1 over
H3K4me2 and H3K4me3 of these Tudor domains.
Based on previous experimental data and our
computational analysis, we conclude that MSH6 and
PDS5C Tudor domains bind to H3K4 preferentially when it is
in the monomethylated state (H3K4me1). Interestingly,
in-silico predictions suggest di
fferent levels of affinity
between the Tudor domains, which could lead to di
fferences
in their contribution to the H3K4me1-associated mutation
rate reduction. The similar binding preference of PDS5C and
MSH6 Tudor domains suggests that multiple repair
pathways could have evolved H3K4me1-targeting potential
in plants, motivating additional experiments and
investigations into the evolutionary origins of these
mechanisms. Because MSH6 operates as a dimer with
MSH2 to form the MutSα complex, which recognizes and
repairs small mismatches, its Tudor domain could explain
the previous observation that MSH2 preferentially targets
gene bodies to reduce mutation rates therein (Bel
field et al.,
2018). These fi
ndings are consistent with extensive work
showing that mutation rates can be lower in gene bodies of
active and conserved genes, but suggest that these
7
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
mechanisms are independent of, while functionally
analogous to, similar mechanisms known in vertebrates
(H3K36me3 targeted repair described in the introduction).
Further experiments are needed - the reduced mutation
rates in gene bodies in active genes in plants may be
explained by multiple mechanisms collectively targeting
H3K4me1 or additional histone states via Tudor domains as
well as other histone modi
fications and readers (Davarinejad
et al., 2022; Liu et al., 2022).
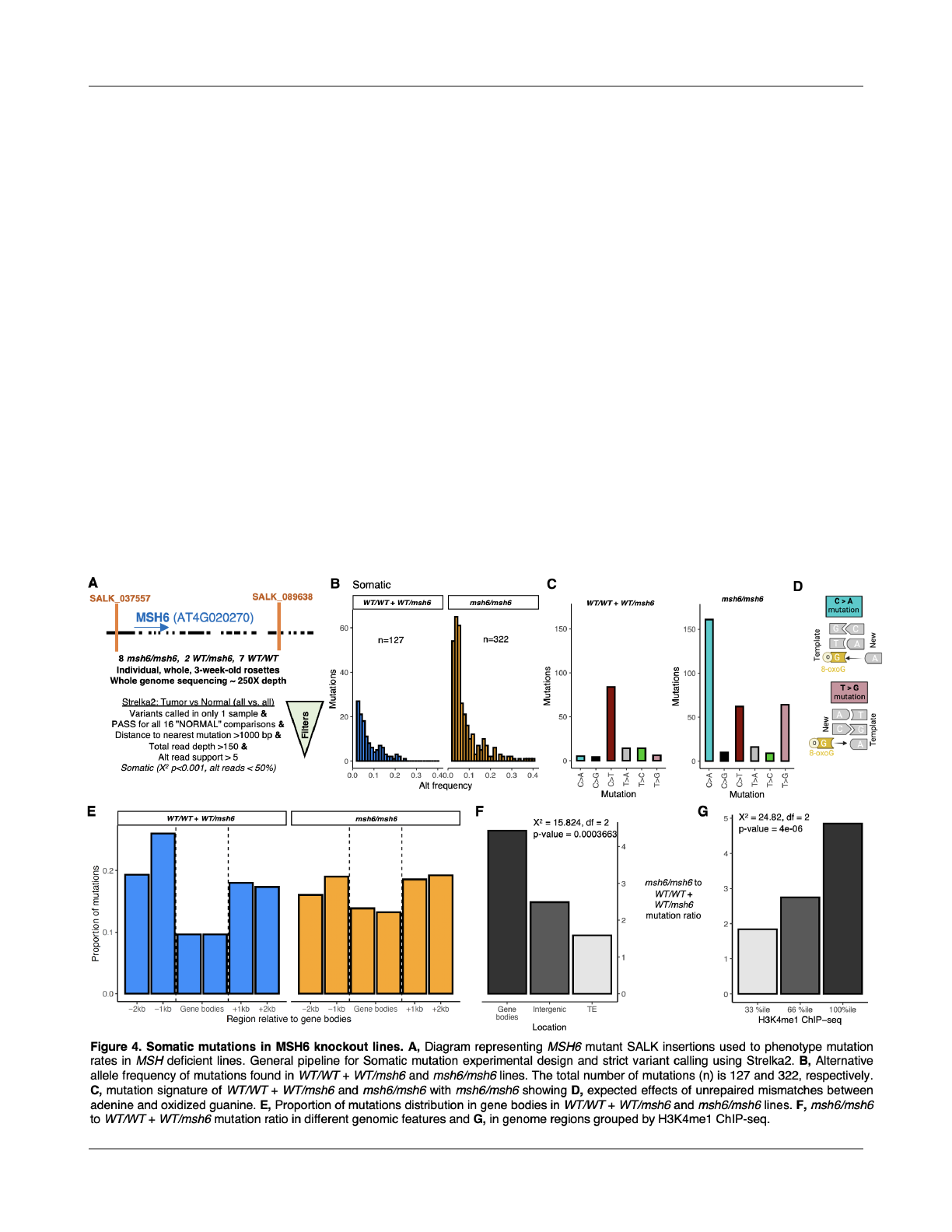
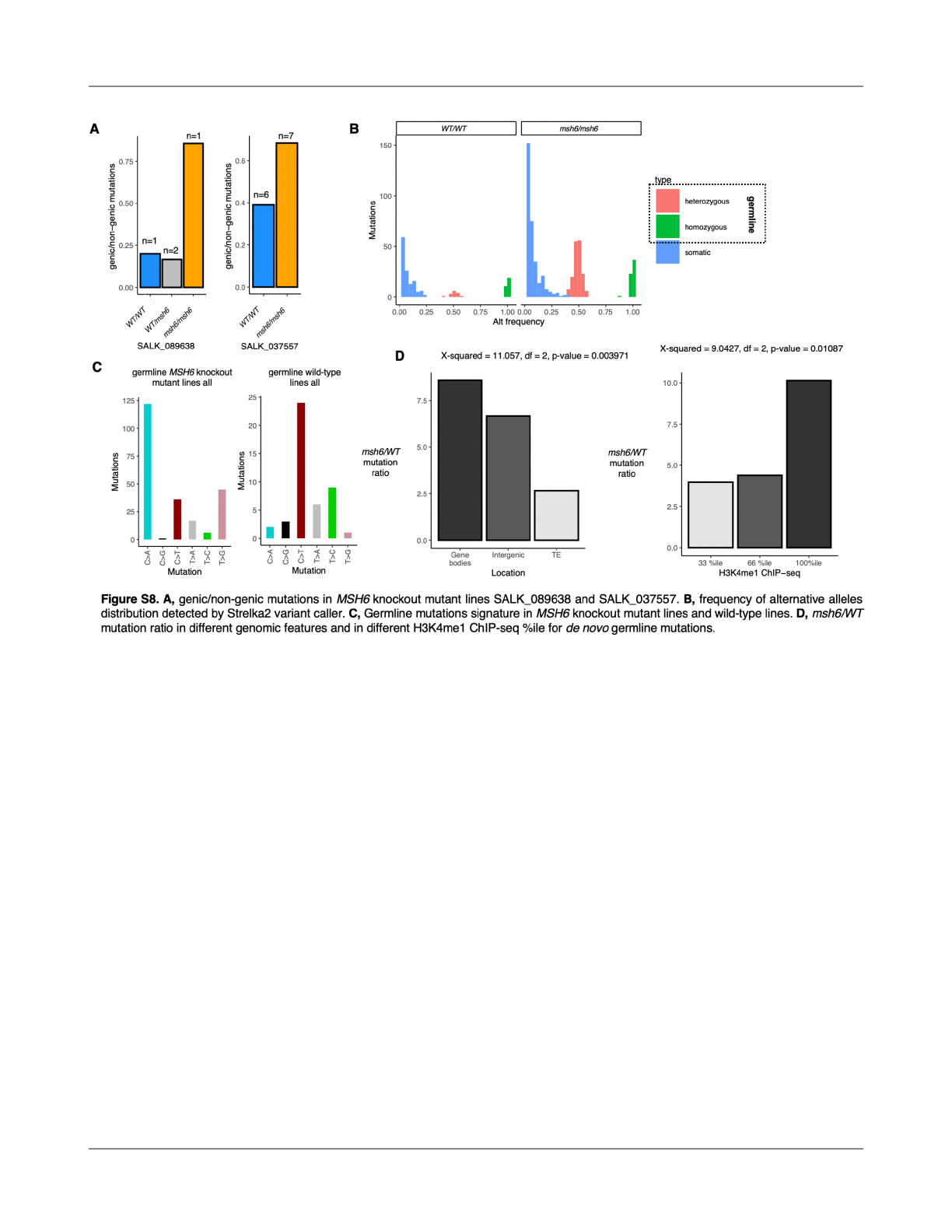
de novo mutations in MSH6 knockout lines indicate
targeted repair of gene bodies and H3K4me1 marked
regions
To experimentally test the e
ffect of MSH6 in
targeted DNA repair and their potential consequence in the
reduction in mutation rates associated with H3K4me1 and
therefore gene bodies, we performed deep sequencing
(250x depth coverage) of 3-week old rosettes of A. thaliana
of 7 MSH6 knockout line SALK_037557 (msh6/msh6), 1
MSH6 knockout line SALK_089638 (msh6/msh6), 2
heterozygous SALK_089638 lines (WT/msh6), and 9
wildtypes (WT/WT) lines (Fig. 4A). Variant caller Strelka2 was
used in TUMOR-NORMAL mode to call SBS mutations for
each sample following a strict fi
ltering process to call true
mutations. In summary, variants were kept if they were
called in only 1 sample, PASS for all 16 “NORMAL”
comparisons reported by Strelka2 (Kim et al., 2018), the
distance to nearest mutation >1000 bp, total read depth
>150, and alt read support > 5. Somatic vs de novo
germline mutations were distinguished based on deviation
from expected alternative variant frequencies (X
2
p<0.001,
alt reads < 50%) (Fig. S8B). As expected, the total number
of somatic mutations was higher in the MSH6 knockout
lines (mean±SE = 40.25±3.14 per sample) than in wild-type
and SALK_089638c heterozygous lines (14.1±2.31 per
sample). Mutations from heterozygotes were combined with
wild-type samples for further comparisons because
heterozygous lines exhibited the wild-type phenotypes (Fig.
S8A).
As expected due to mismatches arising from the
tendency for oxidized guanine (8-oxoG) to mispair with
adenine (Fig. 4D) (Kino et al., 2017), C>A and T>G
substitutions were proportionally higher in MSH6 knockout
lines (Fig. 4C), which is consistent with mutational
signatures observed in mismatch repair (MMR) de
ficient
organisms (Sanders et al., 2021; Lujan et al., 2014). This
increase in C>A and T>G SBS was also found for the de
novo germline mutations (Fig. S6C). This result is consistent
with the high a
ffinity of MutSα for 8-oxoG:A mispairings
(Mazurek et al., 2002). In light of evidence that MSH6
physically binds MutY (Gu et al., 2002; Hahm et al., 2022)
and interacts synergistically with OGG1 (Ni et al., 1999;
Pavlov et al., 2003), both proteins responsible for 8-oxoG:A
base-excision repair (BER), we cannot exclude the
possibility that MSH6 promotes the local activity of multiple
(MMR and BER) repair pathways. MSH6 also in
fluences
somatic recombination in A. thaliana (Li et al., 2006;
Gonzalez and Spampinato, 2020). Future studies should
also consider the potential e
ffects of somatic recombination
on local mutaiton rate.
To test whether MSH6 contributes to the reduction
in mutation rates in gene bodies and H3K4me1 marked
regions, we explored changes in the proportion of mutations
in MSH6 knockout and wild-type lines. The distribution of
mutations in wild-type plants around genes was consistent
with what has been previously reported (Monroe et al.,
2022), while in MSH6 knockout mutant lines gene body
mutation rates were increased, consistent with MSH6
targeting to gene bodies in wild-type plants (Fig. 4E). In
gene bodies, mutation rates were more than 4.5X higher in
MSH6 knockout lines than the wildtype and heterozygous
samples, while the increase of mutations in intergenic
regions and TEs was signi
ficantly less, 2.4X and 1.7X,
respectively (Fig. 4F). We also estimated the mutation ratio
in relation to H3K4me1 ChIP-seq enrichment, and found
that MSH6 knockout lines contain ~5X more mutations in
genome regions most highly enriched for H3K4me1, with
regions containing less H3K4me1 experiencing signi
ficantly
lower increase mutation rates in the knockout lines (Fig. 4G).
8
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
Experimental results are consistent with the
hypothesis that H3K4me1 marked genome regions
experience targeted DNA repair in plants, with the Tudor
domain of repair proteins providing a mechanistic basis for
this recruitment. These results inspire future functional
studies of this and other potential candidate genes
in
fluencing targeted DNA repair and emergent mutation
biases.
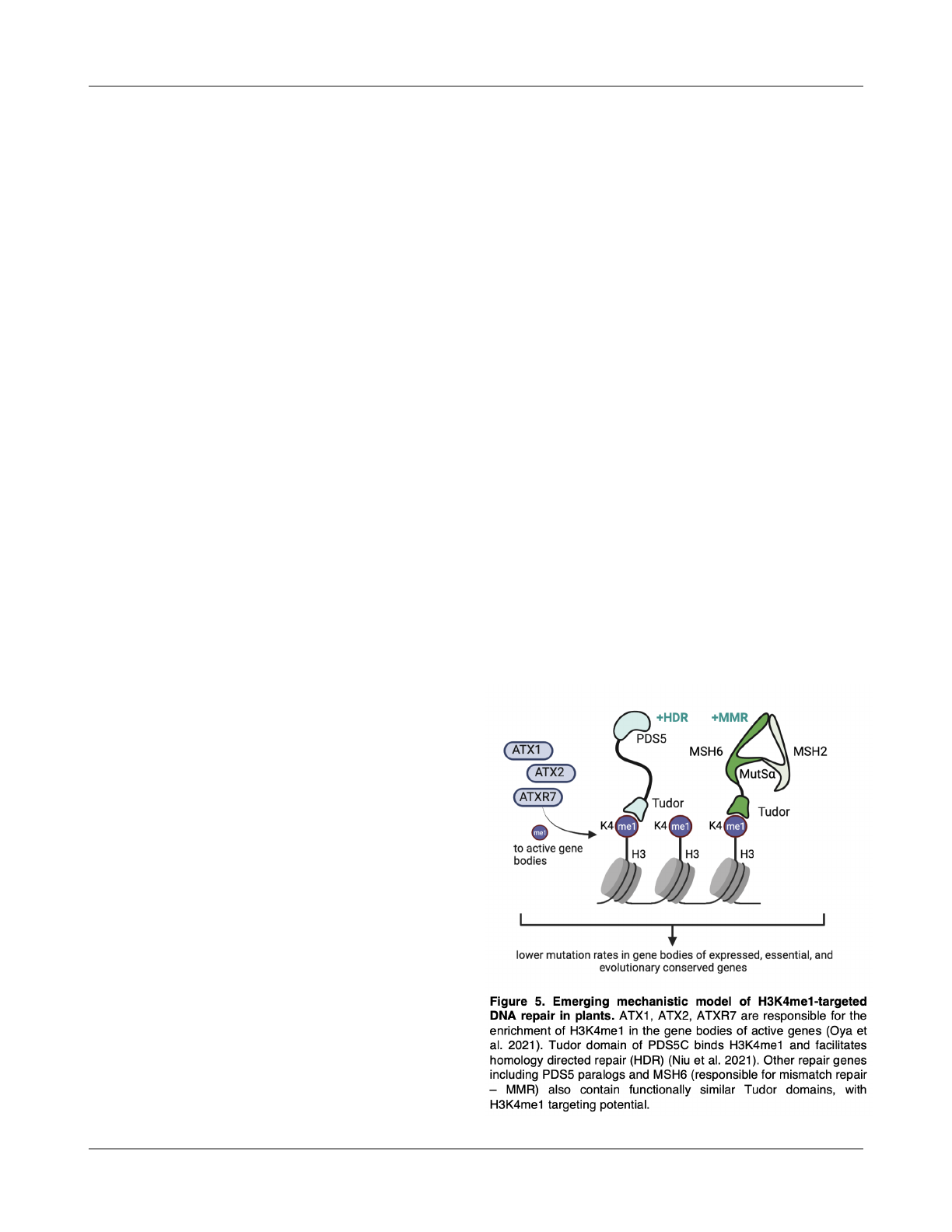
Conclusions
We found evidence of mutation bias associated
with H3K4me1-mediated DNA repair in O. sativa and
examined potential mechanisms conserved in plants (Fig. 5).
Our observations here are derived from reanalyses of data
generated by independent research groups (Li et al., 2017;
Niu et al., 2021; Xie et al., 2021) and prove consistent with
previous reports of mutation biases in A. thaliana (Monroe et
al., 2022). The mechanisms revealed are aligned with
evolutionary models of evolved mutation bias, indicating
targeting of mismatch repair and homology-directed repair
pathways to regions of the genome functionally sensitive to
mutation: coding regions and genes under stronger
evolutionary constraints. Furthermore, we fi
nd genetic
evidence that MSH6 regulates the reduced mutation rate in
gene bodies and H3K4me1-marked regions in A. thaliana.
Functional characterization of PDS5 proteins is still required
to estimate their in
fluence over lower mutation rates in
H3K4me1-associated regions. These fi
ndings provide a
plant-speci
fic and higher-resolution mechanistic model of
hypomutation in gene bodies and essential genes,
motivating experimental investigations to further elucidate
the extent, evolutionary origins, and consequences of
chromatin-targeted DNA repair.
Acknowledgments
The research was conducted at the University of California
Davis, which is located on land which was the home of the
Patwin people for thousands of years. We thank Satoyo
Oya, Tetsuji Kakutani, and Soichi Inagaki for their feedback
and insights on H3K4me1. The Monroe Lab is supported by
FFAR grant ICRC20-0000000014, USDA-NIFA grant
108681-Z5327202, UC Davis STAIR grant, and CPRB grant
HG-2022-35.
Author Contributions
DQ, DL, and GM designed the research; DQ, DL, KZ, LO, AA
and GM performed research; DQ, DL, KZ, AP, VY, PC, and
GM contributed new analytic/computational/etc. tools; DQ,
DL, KZ VY, AP, PC, and GM analyzed data; and DQ, DL, KZ,
VY, PC, and GM wrote the paper.
Methods
Mutation dataset in O. sativa
Germline de novo mutations in 1,504 fast neutron
mutagenesis lines were downloaded from Kitbase at
kitbase.ucdavis.edu. These were independently called and
validated as previously described (Li et al., 2017). We
focused speci
fically on single base substitutions (SBS),
which were validated with a >99% accuracy by Li et al.
(2017). We annotated each SBS in coding regions as being
a synonymous or non-synonymous mutation based on the
e
ffect on the amino acid sequence. We compared
non-synonymous and synonymous ratios with values from
genomes of 3,010 natural accessions (Wang et al., 2018)
and neutral expectations based on mutation spectra, coding
region nucleotide composition, and codon table with the
Null_ns_s function from the polymorphology package in R
(https://github.com/greymonroe/polymorphology/blob/main/
R/Null_ns_s.R).
Epigenomic data collection
Epigenome features were accessed from the RiceENCODE
database (glab.hzau.edu.cn/RiceENCODE/), which has been
previously described (Xie et al., 2021). In brief, peaks were
called from ChIP-seq data with MACS2 (Zhang et al., 2008)
narrow-peak calling settings. We analyzed peak
distributions for H3K4me1, H3K9me1, H3K4me3,
H3K36me3, H3K9me2, H3K27me3, H3K27ac, H3K4ac,
H3K12ac, H3K9ac, and RNA polymerase II (PII) measured in
Nipponbare O. sativa plant seedlings, which constituted the
most complete set of histone modi
fications available. We
repeated analyses but with H3K4me1 measurements
derived from panicles and leaves (rather than seedlings) and
found essentially the same results.
For downstream analyses comparing changes in
mutation in MSH6 knockout mutant lines as a function of
H3K4me1, we accessed a collection of H3K4me1 ChIPseq
datasets from A. thaliana maintained by the Plant Chromatin
State Database (Liu et al., 2018). From these, we calculated
the normalized mean across all datasets in 200 bp sliding
(100 bp) windows across the entire genome.
Estimation of the relationship between mutation rates
and O. sativa epigenomic features
We divided the genome into 100 bp windows
surrounding genes (+- 3000 bp of genes). This allowed us
to, in later steps, restrict our analyses to only genes known
to have accumulated loss-of-function mutations, and thus
be less likely to be a
ffected by selection. We also divided
the genome into 100bp windows and repeated analyses, to
con
firm that results were generally the same. We calculated
9
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
the number of single base-pair substitutions and peaks for
each epigenomic feature overlapping within each window.
We then estimated the relationships between epigenomic
features and mutation rates with a binomial generalized
linear model where the response was a binary state de
fined
as whether a substitution occurred in that window,
predicted by all features, with predictors de
fined as whether
that window overlapped with an epigenome peak. We also
repeated the analyses with a linear regression model where
the response was the number of mutations in a window and
found essentially the same results, so we show the binomial
regression results. To test whether fi
ndings were driven
simply by GC>AT mutations in transposable elements, we
removed all GC>AT and repeated analyses. To further
control for any residual selection in the mutation
accumulation experiment, we also restricted our analyses to
genes harboring loss-of-function mutations in the
population and repeated the analyses. Finally, we restricted
analyses to homozygous SBS and repeated the analyses.
Mutation frequencies were plotted around genes in 100 bp
windows. Since gene bodies are di
fferent lengths, the
position of the window was converted into a percent of
gene length. H3K4me1 peaks around gene bodies were
plotted similarly. We also visualized mutation frequencies
relative to H3K4me1 peaks in the same manner.
Analysis of ChIP-seq data of AtPDS5C
To study the distribution of PDS5C, we used ChIP-seq data
as described by Niu et al. (2021). PDS5C enrichment was
calculated as described by Niu et al. (2021) among regions
as log2[(1 + n_ChIP)/N_ChIP] – log2[(1 + n_Input)/N_Input)],
where n_ChIP and n_Input represent the total depth of
mapped ChIP and Input fragments in a region, and N_ChIP
and N_Input are the numbers total depths of mapped
unique fragments. We calculated PDS5C enrichment in
genic features (1000 bp upstream and downstream of
genes, UTRs, introns, coding regions) and gene bodies (TSS
to TTS) across the TAIR10 A. thaliana genome
(arabidopsis.org).
Relationship between AtPDS5C and functional
constraint
We analyzed the enrichment of the PDS5C ChIP-seq peaks
in A. thaliana in genetic features and estimated the
relationships between those regions and mutation rates,
H4K4me1, Pn/Ps, and tissue expression depth. Tissue
expression data are from (Mergner et al., 2020). H3K4me1 in
Arabidopsis is from the Plant Chromatin State Database (Liu
et al., 2018). Synonymous (Ps) and non-synonymous
polymorphism (Pn) data are from the 1001 Genomes project
(1001 Genomes Consortium, 2016). Essential genes were
based on fi
ndings from (Lloyd and Meinke, 2012). Germline
mutation rates are from (Weng et al., 2019; Monroe et al.,
2022).
Blastp and protein structure prediction and visualization
We used blastp on Phytozome (Goodstein et al., 2012) to
search the O. sativa proteome for PDS5C and MHS6
orthologs and to search the A. thaliana proteome for genes
containing Tudor domains similar to that of PDS5C, which
was validated experimentally to bind H3K4me1 (Niu et al.,
2021). We submitted the resulting list of 29 genes with
putative Tudor domains to gene ontology analysis with
ShinyGO (bioinformatics.sdstate.edu/go/)(Ge et al., 2019).
Protein structure predictions were performed using
AlphaFold (Jumper et al., 2021) in Google Colab (Mirdita et
al., 2022) in no-template mode. All structures were
visualized, processed, and analyzed using UCSF ChimeraX
(Goddard et al., 2018).
Peptide docking
H3 tail peptides comprising 5 amino acids with the different
methylation states for K4 (none, mono, di or trimethylated)
were docked to the experimental structure of A. thaliana
PDS5C Tudor domain and to the models of A. thaliana and
O. sativa MSH6 and O. sativa PDS5C Tudor domains using
Rosetta FlexPepDock tool (Raveh et al., 2010) in re
finement
mode. We generated 10,000 docked models per case and
analyzed the top 10% based on Rosetta's total score.
Analysis of the outputs was conducted using PyRosetta
home-made scripts (Chaudhury et al., 2010) to measure the
average distance of lysine 4 nitrogen in histone 3 to the
center of the aromatic rings in the residues forming the
aromatic cage in Tudor domains.
Somatic mutations in MSH6 Knockout lines
To study the e
ffect of MSH6 in H3K4me1 targeted repair we
performed 250x coverage sequencing analyses of 17 A.
thaliana; 9 WT/WT, 7 msh6/msh6 (6 SALK_037557 and 1
SALK_089638) and 2 msh6/WT (2 heterozygous
SALK_089638). In brief, 3 week old plants were harvested,
stems and roots were removed and rosettes were stored at
-80 ºC. Whole rosettes were grinded using a mortar and
liquid nitrogen and Qiagen DNeasy Plant Mini Kit (Cat No:
69106) following the manufacturer's instructions. Illumina
library preparation and Whole-genome DNA sequencing
with NovaSeq (150-bp read length at high genomic
coverage ~250x) was performed by the UC Davis Genome
Center. Reads were trimmed with trimmomatic (Bolger et
al., 2014) and duplicates were marked with the samtools
markdup function (Li et al., 2009). Reads were mapped to
10
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
the A. thaliana TAIR10 reference genome with bwa mem(Li
and Durbin, 2009). Strelka2 variant caller in
TUMOR-NORMAL mode (each sample was compared to all
other 16 samples as NORMALs) was used to call SBS in the
sequenced samples (Kim et al., 2018). Mutations found
were fi
ltered by keeping variants called in only one sample,
given a PASS quality score in all 16 ”NORMAL”
comparisons. In addition to the built-in fi
lters imposed by
Strelka2 for variants to receive a PASS, which “include (1)
the genotype probability computed by the core variant
probability model, (2) root-mean-square mapping quality, (3)
strand bias, (4) the fraction of reads consistent with locus
haplotype model, and (5) the complexity of the reference
context as measured by metrics such as homopolymer
length and compressibility” (Kim et al., 2018), we further
fi
ltered to only include variants in which the distance to the
nearest mutation was >1000 bp, total read depth >150 and
alt read support > 5. To distinguish somatic mutations from
de novo heterozygous variants, read-depths between
alternative and reference alleles were compared with an X
2
test against the expected 50/50 ratio for heterozygous
variants. Mutations were considered somatic if the X
2
test
statistic p-value was less than 0.001 (to account for
multiple testing) and the alt read depth was < 50%
Homozygous de novo germline mutations were identi
fied
when X
2
test statistic p-value was less than 0.001 and the
alt read depth was >50%. Complete code for variant calling,
post-processing, and analyses are maintained on
https://github.com/greymonroe/Quiroz_H3K4me1_mediated
_repair.
Code and data
Figures, code, and data are located on:
https://github.com/greymonroe/Quiroz_H3K4me1_mediated
_repair.
References
1001 Genomes Consortium (2016). 1,135 Genomes Reveal
the Global Pattern of Polymorphism in Arabidopsis
thaliana. Cell 166: 481–491.
Adé, J., Belzile, F., Philippe, H., and Doutriaux, M.P.
(1999). Four mismatch repair paralogues coexist in
Arabidopsis thaliana: AtMSH2, AtMSH3, AtMSH6-1 and
AtMSH6-2. Mol. Gen. Genet. 262: 239–249.
Akdemir, K.C. et al. (2020). Somatic mutation distributions
in cancer genomes vary with three-dimensional
chromatin structure. Nat. Genet. 52: 1178–1188.
Aymard, F., Bugler, B., Schmidt, C.K., Guillou, E., Caron,
P., Briois, S., Iacovoni, J.S., Daburon, V., Miller, K.M.,
Jackson, S.P., and Legube, G. (2014). Transcriptionally
active chromatin recruits homologous recombination at
DNA double-strand breaks. Nat. Struct. Mol. Biol. 21:
366–374.
Bel
field, E.J., Brown, C., Ding, Z.J., Chapman, L., Luo,
M., Hinde, E., van Es, S.W., Johnson, S., Ning, Y.,
Zheng, S.J., Mithani, A., and Harberd, N.P. (2021).
Thermal stress accelerates Arabidopsis thaliana
mutation rate. Genome Res. 31: 40–50.
Bel
field, E.J., Ding, Z.J., Jamieson, F.J.C., Visscher, A.M.,
Zheng, S.J., Mithani, A., and Harberd, N.P. (2018).
DNA mismatch repair preferentially protects genes from
mutation. Genome Res. 28: 66–74.
Bolaños-Villegas, P., Yang, X., Wang, H.-J., Juan, C.-T.,
Chuang, M.-H., Makaro
ff, C.A., and Jauh, G.-Y.
(2013). Arabidopsis CHROMOSOME TRANSMISSION
FIDELITY 7 (AtCTF7/ECO1) is required for DNA repair,
mitosis and meiosis. Plant J. 75: 927–940.
Bolger, A.M., Lohse, M., and Usadel, B. (2014).
Trimmomatic: a
flexible trimmer for Illumina sequence
data. Bioinformatics 30: 2114–2120.
Cagan, A. et al. (2022). Somatic mutation rates scale with
lifespan across mammals. Nature 604: 517–524.
Chaudhury, S., Lyskov, S., and Gray, J.J. (2010).
PyRosetta: a script-based interface for implementing
molecular modeling algorithms using Rosetta.
Bioinformatics 26: 689–691.
Davarinejad, H. et al. (2022). The histone H3.1 variant
regulates TONSOKU-mediated DNA repair during
replication. Science 375: 1281–1286.
Fang, H., Zhu, X., Yang, H., Oh, J., Barbour, J.A., and
Wong, J.W.H. (2021). De
ficiency of
replication-independent DNA mismatch repair drives a
5-methylcytosine deamination mutational signature in
cancer. Sci Adv 7: eabg4398.
Foster, P.L., Lee, H., Popodi, E., Townes, J.P., and Tang,
H. (2015). Determinants of spontaneous mutation in the
bacterium Escherichia coli as revealed by
whole-genome sequencing. Proc. Natl. Acad. Sci. U. S.
A. 112: E5990–9.
Ge, S.X., Jung, D., and Yao, R. (2019). ShinyGO: a
graphical gene-set enrichment tool for animals and
plants. Bioinformatics 36: 2628–2629.
Goddard, T.D., Huang, C.C., Meng, E.C., Pettersen, E.F.,
Couch, G.S., Morris, J.H., and Ferrin, T.E. (2018).
UCSF ChimeraX: Meeting modern challenges in
visualization and analysis. Protein Sci. 27: 14–25.
Gonzalez, V. and Spampinato, C.P. (2020). The mismatch
repair protein MSH6 regulates somatic recombination in
Arabidopsis thaliana. DNA Repair 87: 102789.
11
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
Goodstein, D.M., Shu, S., Howson, R., Neupane, R.,
Hayes, R.D., Fazo, J., Mitros, T., Dirks, W., Hellsten,
U., Putnam, N., and Rokhsar, D.S. (2012). Phytozome:
a comparative platform for green plant genomics.
Nucleic Acids Res. 40: D1178–86.
Gu, Y., Parker, A., Wilson, T.M., Bai, H., Chang, D.-Y., and
Lu, A.-L. (2002). Human MutY Homolog, a DNA
Glycosylase Involved in Base Excision Repair,
Physically and Functionally Interacts with Mismatch
Repair Proteins Human MutS Homolog 2/Human MutS
Homolog 6*. J. Biol. Chem. 277: 11135–11142.
Habig, M., Lorrain, C., Feurtey, A., Komluski, J., and
Stukenbrock, E.H. (2021). Epigenetic modi
fications
a
ffect the rate of spontaneous mutations in a
pathogenic fungus. Nat. Commun. 12: 5869.
Hahm, J.Y., Park, J., Jang, E.-S., and Chi, S.W. (2022).
8-Oxoguanine: from oxidative damage to epigenetic
and epitranscriptional modi
fication. Exp. Mol. Med. 54:
1626–1642.
Hill, V.K., Kim, J.-S., and Waldman, T. (2016). Cohesin
mutations in human cancer. Biochim. Biophys. Acta
1866: 1–11.
Huang, Y., Gu, L., and Li, G.-M. (2018).
H3K36me3-mediated mismatch repair preferentially
protects actively transcribed genes from mutation. J.
Biol. Chem. 293: 7811–7823.
Jiang, P., Ollodart, A.R., Sudhesh, V., Herr, A.J., Dunham,
M.J., and Harris, K. (2021). A modi
fied fluctuation
assay reveals a natural mutator phenotype that drives
mutation spectrum variation within Saccharomyces
cerevisiae. Elife 10.
Jumper, J. et al. (2021). Highly accurate protein structure
prediction with AlphaFold. Nature 596: 583–589.
Katju, V., Konrad, A., Deiss, T.C., and Bergthorsson, U.
(2022). Mutation rate and spectrum in obligately
outcrossing Caenorhabditis elegans mutation
accumulation lines subjected to RNAi-induced
knockdown of the mismatch repair gene msh-2. G3 12:
jkab364.
Kawahara, Y. et al. (2013). Improvement of the Oryza sativa
Nipponbare reference genome using next generation
sequence and optical map data. Rice 6: 4.
Kim, J., Daniel, J., Espejo, A., Lake, A., Krishna, M., Xia,
L., Zhang, Y., and Bedford, M.T. (2006). Tudor, MBT
and chromo domains gauge the degree of lysine
methylation. EMBO Rep. 7: 397–403.
Kim, S., Sche
ffler, K., Halpern, A.L., Bekritsky, M.A., Noh,
E., Källberg, M., Chen, X., Kim, Y., Beyter, D.,
Krusche, P., and Saunders, C.T. (2018). Strelka2: fast
and accurate calling of germline and somatic variants.
Nat. Methods 15: 591–594.
Kino, K., Hirao-Suzuki, M., Morikawa, M., Sakaga, A.,
and Miyazawa, H. (2017). Generation, repair and
replication of guanine oxidation products. Genes
Environ 39: 21.
Kolodner, R. (1996). Biochemistry and genetics of
eukaryotic mismatch repair. Genes Dev. 10: 1433–1442.
Krasovec, M., Eyre-Walker, A., Sanchez-Ferandin, S.,
and Piganeau, G. (2017). Spontaneous Mutation Rate
in the Smallest Photosynthetic Eukaryotes. Mol. Biol.
Evol. 34: 1770–1779.
Li, F., Mao, G., Tong, D., Huang, J., Gu, L., Yang, W., and
Li, G.-M. (2013). The histone mark H3K36me3 regulates
human DNA mismatch repair through its interaction with
MutSα. Cell 153: 590–600.
Li, G. et al. (2017). The Sequences of 1504 Mutants in the
Model Rice Variety Kitaake Facilitate Rapid Functional
Genomic Studies. Plant Cell 29: 1218–1231.
Li, H. and Durbin, R. (2009). Fast and accurate short read
alignment with Burrows–Wheeler transform.
Bioinformatics 25: 1754–1760.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J.,
Homer, N., Marth, G., Abecasis, G., and Durbin, R.
(2009). The Sequence Alignment/Map format and
SAMtools. Bioinformatics 25: 2078–2079.
Li, L., Jean, M., and Belzile, F. (2006). The impact of
sequence divergence and DNA mismatch repair on
homeologous recombination in Arabidopsis. Plant J. 45:
908–916.
Li, R. et al. (2021). A body map of somatic mutagenesis in
morphologically normal human tissues. Nature 597:
398–403.
Liu, H. and Zhang, J. (2022). Is the Mutation Rate Lower in
Genomic Regions of Stronger Selective Constraints?
Mol. Biol. Evol. 39.
Liu, Q., Liu, P., Ji, T., Zheng, L., Shen, C., Ran, S., Liu, J.,
Zhao, Y., Niu, Y., Wang, T., and Dong, J. (2022). The
histone methyltransferase SUVR2 promotes DSB repair
via chromatin remodeling and liquid-liquid phase
separation. Mol. Plant.
Liu, Y., Tian, T., Zhang, K., You, Q., Yan, H., Zhao, N., Yi,
X., Xu, W., and Su, Z. (2018). PCSD: a plant chromatin
state database. Nucleic Acids Res. 46: D1157–D1167.
Lloyd, J. and Meinke, D. (2012). A comprehensive dataset
of genes with a loss-of-function mutant phenotype in
Arabidopsis. Plant Physiol. 158: 1115–1129.
Lloyd, J.P., Seddon, A.E., Moghe, G.D., Simenc, M.C.,
12
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
and Shiu, S.-H. (2015). Characteristics of Plant
Essential Genes Allow for within- and between-Species
Prediction of Lethal Mutant Phenotypes. Plant Cell 27:
2133–2147.
López-Cortegano, E., Craig, R.J., Chebib, J., Samuels, T.,
Morgan, A.D., Kraemer, S.A., Böndel, K.B., Ness,
R.W., Colegrave, N., and Keightley, P.D. (2021). De
Novo Mutation Rate Variation and Its Determinants in
Chlamydomonas. Mol. Biol. Evol. 38: 3709–3723.
Lujan, S.A., Clausen, A.R., Clark, A.B., MacAlpine, H.K.,
MacAlpine, D.M., Malc, E.P., Mieczkowski, P.A.,
Burkholder, A.B., Fargo, D.C., Gordenin, D.A., and
Kunkel, T.A. (2014). Heterogeneous polymerase
fidelity
and mismatch repair bias genome variation and
composition. Genome Res. 24: 1751.
Lu, R. and Wang, G.G. (2013). Tudor: a versatile family of
histone methylation “readers.” Trends Biochem. Sci. 38:
546–555.
Lu, Z., Cui, J., Wang, L., Teng, N., Zhang, S., Lam, H.-M.,
Zhu, Y., Xiao, S., Ke, W., Lin, J., Xu, C., and Jin, B.
(2021). Genome-wide DNA mutations in Arabidopsis
plants after multigenerational exposure to high
temperatures. Genome Biol. 22: 160.
Lynch, M. (2010). Evolution of the mutation rate. Trends
Genet. 26: 345–352.
Lynch, M., Ackerman, M.S., Gout, J.-F., Long, H., Sung,
W., Thomas, W.K., and Foster, P.L. (2016). Genetic
drift, selection and the evolution of the mutation rate.
Nat. Rev. Genet. 17: 704–714.
Makova, K.D. and Hardison, R.C. (2015). The e
ffects of
chromatin organization on variation in mutation rates in
the genome. Nat. Rev. Genet. 16: 213–223.
Martincorena, I. and Luscombe, N.M. (2013). Non-random
mutation: the evolution of targeted hypermutation and
hypomutation. Bioessays 35: 123–130.
Maurer-Stroh, S., Dickens, N.J., Hughes-Davies, L.,
Kouzarides, T., Eisenhaber, F., and Ponting, C.P.
(2003). The Tudor domain “Royal Family”: Tudor, plant
Agenet, Chromo, PWWP and MBT domains. Trends
Biochem. Sci. 28: 69–74.
Mazurek, A., Berardini, M., and Fishel, R. (2002).
Activation of Human MutS Homologs by 8-Oxo-guanine
DNA Damage*. J. Biol. Chem. 277: 8260–8266.
Mergner, J. et al. (2020). Mass-spectrometry-based draft of
the Arabidopsis proteome. Nature 579: 409–414.
Mirdita, M., Schütze, K., Moriwaki, Y., Heo, L.,
Ovchinnikov, S., and Steinegger, M. (2022).
ColabFold: making protein folding accessible to all. Nat.
Methods 19: 679–682.
Monroe, J.G. et al. (2022). Mutation bias re
flects natural
selection in Arabidopsis thaliana. Nature.
Moore, L. et al. (2021). The mutational landscape of human
somatic and germline cells. Nature.
Morales, C., Ruiz-Torres, M., Rodríguez-Acebes, S.,
Lafarga, V., Rodríguez-Corsino, M., Megías, D.,
Cisneros, D.A., Peters, J.-M., Méndez, J., and
Losada, A. (2020). PDS5 proteins are required for
proper cohesin dynamics and participate in replication
fork protection. J. Biol. Chem. 295: 146–157.
Ni, T.T., Marsischky, G.T., and Kolodner, R.D. (1999).
MSH2 and MSH6 are required for removal of adenine
misincorporated opposite 8-oxo-guanine in S.
cerevisiae. Mol. Cell 4: 439–444.
Niu, Q. et al. (2021). A histone H3K4me1-speci
fic binding
protein is required for siRNA accumulation and DNA
methylation at a subset of loci targeted by
RNA-directed DNA methylation. Nat. Commun. 12:
3367.
Oya, S., Takahashi, M., Takashima, K., Kakutani, T., and
Inagaki, S. (2021). Transcription-coupled and
epigenome-encoded mechanisms direct H3K4
methylation. bioRxiv: 2021.06.03.446702.
Pavlov, Y.I., Mian, I.M., and Kunkel, T.A. (2003). Evidence
for preferential mismatch repair of lagging strand DNA
replication errors in yeast. Curr. Biol. 13: 744–748.
de la Peña, M.V., Summanen, P.A.M., Liukkonen, M., and
Kronholm, I. (2022). Chromatin structure in
fluences
rate and spectrum of spontaneous mutations in
Neurospora crassa. bioRxiv: 2022.03.13.484164.
Phipps, J. and Dubrana, K. (2022). DNA Repair in Space
and Time: Safeguarding the Genome with the Cohesin
Complex. Genes 13.
Pradillo, M., Knoll, A., Oliver, C., Varas, J., Corredor, E.,
Puchta, H., and Santos, J.L. (2015). Involvement of the
Cohesin Cofactor PDS5 (SPO76) During Meiosis and
DNA Repair in Arabidopsis thaliana. Front. Plant Sci. 6:
1034.
Raveh, B., London, N., and Schueler-Furman, O. (2010).
Sub-angstrom modeling of complexes between
flexible
peptides and globular proteins. Proteins 78: 2029–2040.
Ren, Q., Yang, H., Rosinski, M., Conrad, M.N., Dresser,
M.E., Guacci, V., and Zhang, Z. (2005). Mutation of the
cohesin related gene PDS5 causes cell death with
predominant apoptotic features in Saccharomyces
cerevisiae during early meiosis. Mutat. Res. 570:
163–173.
Sanders, M.A. et al. (2021). Life without mismatch repair.
bioRxiv: 2021.04.14.437578.
13
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
Sasani, T.A., Ashbrook, D.G., Beichman, A.C., Lu, L.,
Palmer, A.A., Williams, R.W., Pritchard, J.K., and
Harris, K. (2022). A natural mutator allele shapes
mutation spectrum variation in mice. Nature 605:
497–502.
Schep, R. et al. (2021). Impact of chromatin context on
Cas9-induced DNA double-strand break repair pathway
balance. Mol. Cell 81: 2216–2230.e10.
Schubert, V., Weissleder, A., Ali, H., Fuchs, J.,
Lermontova, I., Meister, A., and Schubert, I. (2009).
Cohesin gene defects may impair sister chromatid
alignment and genome stability in Arabidopsis thaliana.
Chromosoma 118: 591–605.
Schuster-Böckler, B. and Lehner, B. (2012). Chromatin
organization is a major in
fluence on regional mutation
rates in human cancer cells. Nature 488: 504–507.
Sun, Z., Zhang, Y., Jia, J., Fang, Y., Tang, Y., Wu, H., and
Fang, D. (2020). H3K36me3, message from chromatin
to DNA damage repair. Cell Biosci. 10: 9.
Supek, F. and Lehner, B. (2017). Clustered Mutation
Signatures Reveal that Error-Prone DNA Repair Targets
Mutations to Active Genes. Cell 170: 534–547.e23.
Supek, F. and Lehner, B. (2015). Di
fferential DNA mismatch
repair underlies mutation rate variation across the
human genome. Nature 521: 81–84.
Supek, F. and Lehner, B. (2019). Scales and mechanisms
of somatic mutation rate variation across the human
genome. DNA Repair 81: 102647.
Wang, W. et al. (2018). Genomic variation in 3,010 diverse
accessions of Asian cultivated rice. Nature 557: 43–49.
Weiss, T., Crisp, P.A., Rai, K.M., Song, M., Springer, N.M.,
and Zhang, F. (2022). Drastic di
fferential CRISPR-Cas9
induced mutagenesis in
fluenced by DNA methylation
and chromatin features. bioRxiv: 2022.02.28.482333.
Weng, M.-L., Becker, C., Hildebrandt, J., Neumann, M.,
Rutter, M.T., Shaw, R.G., Weigel, D., and Fenster,
C.B. (2019). Fine-Grained Analysis of Spontaneous
Mutation Spectrum and Frequency in Arabidopsis
thaliana. Genetics 211: 703–714.
Wyant, S.R., Rodriguez, M.F., Carter, C.K., Parrott, W.A.,
Jackson, S.A., Stupar, R.M., and Morrell, P.L. (2022).
Fast neutron mutagenesis in soybean enriches for small
indels and creates frameshift mutations. G3 12.
Xie, L., Liu, M., Zhao, L., Cao, K., Wang, P., Xu, W., Sung,
W.-K., Li, X., and Li, G. (2021). RiceENCODE: A
comprehensive epigenomic database as a rice
Encyclopedia of DNA Elements. Mol. Plant 14:
1604–1606.
Yang, X. et al. (2021). Developmental and temporal
characteristics of clonal sperm mosaicism. Cell 184:
4772–4783.e15.
Yan, W., Deng, X.W., Yang, C., and Tang, X. (2021). The
Genome-Wide EMS Mutagenesis Bias Correlates With
Sequence Context and Chromatin Structure in Rice.
Front. Plant Sci. 12: 579675.
Zhang, Y., Liu, T., Meyer, C.A., Eeckhoute, J., Johnson,
D.S., Bernstein, B.E., Nusbaum, C., Myers, R.M.,
Brown, M., Li, W., and Liu, X.S. (2008). Model-based
analysis of ChIP-Seq (MACS). Genome Biol. 9: R137.
Zhu, X., Xie, S., Tang, K., Kalia, R.K., Liu, N., Ma, J.,
Bressan, R.A., and Zhu, J.-K. (2021). Non-CG DNA
methylation-de
ficiency mutations enhance mutagenesis
rates during salt adaptation in cultured Arabidopsis
cells. Stress Biology 1: 12.
14
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
15
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
16
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
17
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
18
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
19
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
20
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint

Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
21
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
22
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
23
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
Quiroz et al., 2022, biorxiv H3K4me1-targeted DNA repair in plants
Supplemental Table 1
Enrichment FDR
nGenes
Pathway Genes
Fold Enrichment
Pathway
URL
Genes
4.04E-16
7
25
483.49
Mitotic sister
chromatid
cohesion
http://amigo.gene
ontology.org/amig
o/term/GO:00070
64
AT1G15940
AT1G77600
AT1G80810
AT4G31880
AT4G32620
AT5G10950
AT5G47690
3.08E-14
7
48
251.817708
Sister chromatid
cohesion
http://amigo.gene
ontology.org/amig
o/term/GO:00070
62
AT1G15940
AT1G77600
AT1G80810
AT4G31880
AT4G32620
AT5G10950
AT5G47690
3.70E-06
3
26
199.240385
Microsporogenesi
s
http://amigo.gene
ontology.org/amig
o/term/GO:00095
56
AT1G15940
AT4G31880
AT5G47690
1.21E-12
7
87
138.933908
Sister chromatid
segregation
http://amigo.gene
ontology.org/amig
o/term/GO:00008
19
AT1G15940
AT1G77600
AT1G80810
AT4G31880
AT4G32620
AT5G10950
AT5G47690
4.08E-12
7
109
110.892202
Mitotic nuclear
division
http://amigo.gene
ontology.org/amig
o/term/GO:01400
14
AT1G15940
AT1G77600
AT1G80810
AT4G31880
AT4G32620
AT5G10950
AT5G47690
8.62E-12
7
126
95.9305556
Nuclear
chromosome
segregation
http://amigo.gene
ontology.org/amig
o/term/GO:00988
13
AT1G15940
AT1G77600
AT1G80810
AT4G31880
AT4G32620
AT5G10950
AT5G47690
1.97E-11
7
146
82.7893836
Chromosome
segregation
http://amigo.gene
ontology.org/amig
o/term/GO:00070
59
AT1G15940
AT1G77600
AT1G80810
AT4G31880
AT4G32620
AT5G10950
AT5G47690
2.95E-10
7
217
55.7016129
Nuclear division
http://amigo.gene
ontology.org/amig
o/term/GO:00002
80
AT1G15940
AT1G77600
AT1G80810
AT4G31880
AT4G32620
AT5G10950
AT5G47690
5.37E-10
7
242
49.947314
Mitotic cell cycle
proc.
http://amigo.gene
ontology.org/amig
o/term/GO:19030
47
AT1G15940
AT1G77600
AT1G80810
AT4G31880
AT4G32620
AT5G10950
AT5G47690
8.03E-10
7
259
46.6689189
Organelle fission
http://amigo.gene
ontology.org/amig
o/term/GO:00482
85
AT1G15940
AT1G77600
AT1G80810
AT4G31880
AT4G32620
AT5G10950
AT5G47690
6.42E-12
9
397
39.145466
DNA repair
http://amigo.gene
ontology.org/amig
o/term/GO:00062
81
AT1G15940
AT1G77600
AT1G80810
AT4G02070
AT4G31880
AT4G32620
AT4G32970
AT5G10950
AT5G47690
1.05E-11
9
431
36.0574246
Cellular response
to DNA damage
stimulus
http://amigo.gene
ontology.org/amig
o/term/GO:00069
74
AT1G15940
AT1G77600
AT1G80810
AT4G02070
AT4G31880
AT4G32620
AT4G32970
AT5G10950
AT5G47690
3.00E-12
10
564
30.6161348
Chromosome
organization
http://amigo.gene
ontology.org/amig
o/term/GO:00512
76
AT1G05830
AT1G15940
AT1G77600
AT1G80810
AT2G31650
AT4G02070
AT4G31880
AT4G32620
AT5G10950
AT5G47690
1.14E-09
8
484
28.5413223
Cell cycle proc.
http://amigo.gene
ontology.org/amig
o/term/GO:00224
02
AT1G15940
AT1G77600
AT1G80810
AT4G02070
AT4G31880
AT4G32620
AT5G10950
AT5G47690
4.51E-10
9
678
22.9214602
DNA metabolic
proc.
http://amigo.gene
ontology.org/amig
o/term/GO:00062
59
AT1G15940
AT1G77600
AT1G80810
AT4G02070
AT4G31880
AT4G32620
AT4G32970
AT5G10950
AT5G47690
7.35E-08
9
1265
12.2851779
Cellular response
to stress
http://amigo.gene
ontology.org/amig
o/term/GO:00335
54
AT1G15940
AT1G77600
AT1G80810
AT4G02070
AT4G31880
AT4G32620
AT4G32970
AT5G10950
AT5G47690
9.00E-08
10
1845
9.35907859
Organelle
organization
http://amigo.gene
ontology.org/amig
o/term/GO:00069
96
AT1G05830
AT1G15940
AT1G77600
AT1G80810
AT2G31650
AT4G02070
AT4G31880
AT4G32620
AT5G10950
AT5G47690
24
.CC-BY 4.0 International licenseavailable under a
(which was not certified by peer review) is the author/funder, who has granted bioRxiv a license to display the preprint in perpetuity. It is made
The copyright holder for this preprintthis version posted November 11, 2022. ; https://doi.org/10.1101/2022.05.28.493846doi: bioRxiv preprint
