
Guide to Interpreting
Genomic Reports:
A Genomics Toolkit
A guide to genomic test results for
non-genetics providers
Created by the Practitioner Education Working
Group of the Clinical Sequencing Exploratory
Research (CSER) Consortium
Authors
Kelly East, MS, CGC, Wendy Chung MD, PhD, Kate Foreman, MS, CGC,
Mari Gilmore, MS, CGC, Michele Gornick, PhD, Lucia Hindor, PhD, Tia Kauman,
MPH, Donna Messersmith , PhD, Cindy Prows, MSN, APRN, CNS, Elena Stoel, MD,
Joon-Ho Yu, MPh, PhD and Sharon Plon, MD, PhD
About this resource
This resource was created by a team of genomic testing experts. It is designed to
help non-geneticist healthcare providers to understand genomic medicine and
genome sequencing. The CSER Consortium
1
is an NIH-funded group exploring
genomic testing in clinical settings.
Acknowledgements
This work was conducted as part of the Clinical Sequencing Exploratory Research
(CSER) Consortium, grants U01 HG006485, U01 HG006485, U01 HG006546, U01
HG006492, UM1 HG007301, UM1 HG007292, UM1 HG006508, U01 HG006487,
U01 HG006507, R01 HG006618, and U01 HG007307. Special thanks to Alexandria
Wyatt and Hugo O’Campo for graphic design and layout, Jill Pope for technical
editing, and the entire CSER Practitioner Education Working Group for their time,
energy, and support in developing this resource.
Genomic Report Toolkit

Contents
1
3
8
10
13
15
19
23
26
29
Introduction and Overview ................................................................
Uncertain Results Related to Patient Symptoms: Variants of Uncertain Signicance . . . . . . . . . .
Medically Actionable Secondary Results ....................................................
Diagnostic Results Related to Patient Symptoms: Pathogenic and Likely Pathogenic Variants . . .
No Findings Related to Patient Symptoms ....................................................
Common Risk Allele Results ................................................................
Carrier Status Results ......................................................................
Pharmacogenetic Results ..................................................................
Looking Forward ...........................................................................
3
5
7
2
4
6
8
9
3
1: Introduction and Overview
Key Points:
• The use of genomic tests (gene panels, exome sequencing, genome sequencing) is increasing
in the clinical and research setting.
• An individual’s genetic code contains millions of dierences when compared to the human
reference sequence. These dierences, referred to here as variants are also sometimes called
mutations.
• Genetic variants may be benign and have no impact or may be pathogenic and causative of
disease. When it is unclear whether a variant has an impact it is referred to as a variant of
uncertain signicance.
• Genomic tests are often performed to make a diagnosis and explain symptoms. Results
related to symptoms are called primary ndings while results that are unrelated to symptoms
are called secondary ndings.
• Results of genomic testing may have medical and personal value to both the individual who
underwent testing as well as his relatives.
This guide is intended for healthcare providers faced with understanding and interpreting their
patients’ genomic test reports. This guide is not intended to help select a test, but rather to help
providers navigate test results.
Genetic Tests vs. Genomic Tests
Genetic testing allows for the identication of changes in chromosomes, genes, or proteins (a
genes encoded product). The results can conrm or rule out a suspected genetic condition or help
determine a person’s chance of developing or passing on a genetic disorder.
While genetic testing has been performed for decades, over the past few years there has been a
tremendous increase in the number and scope of genetic tests ordered due to improvements in
technology and decreases in cost. Genomic tests that explore multiple genes (panels), most genes,
and even tests that explore a person’s entire genome, have become a reality. Yet, many healthcare
providers have not been trained in how to understand the output of these increasingly common
tests.
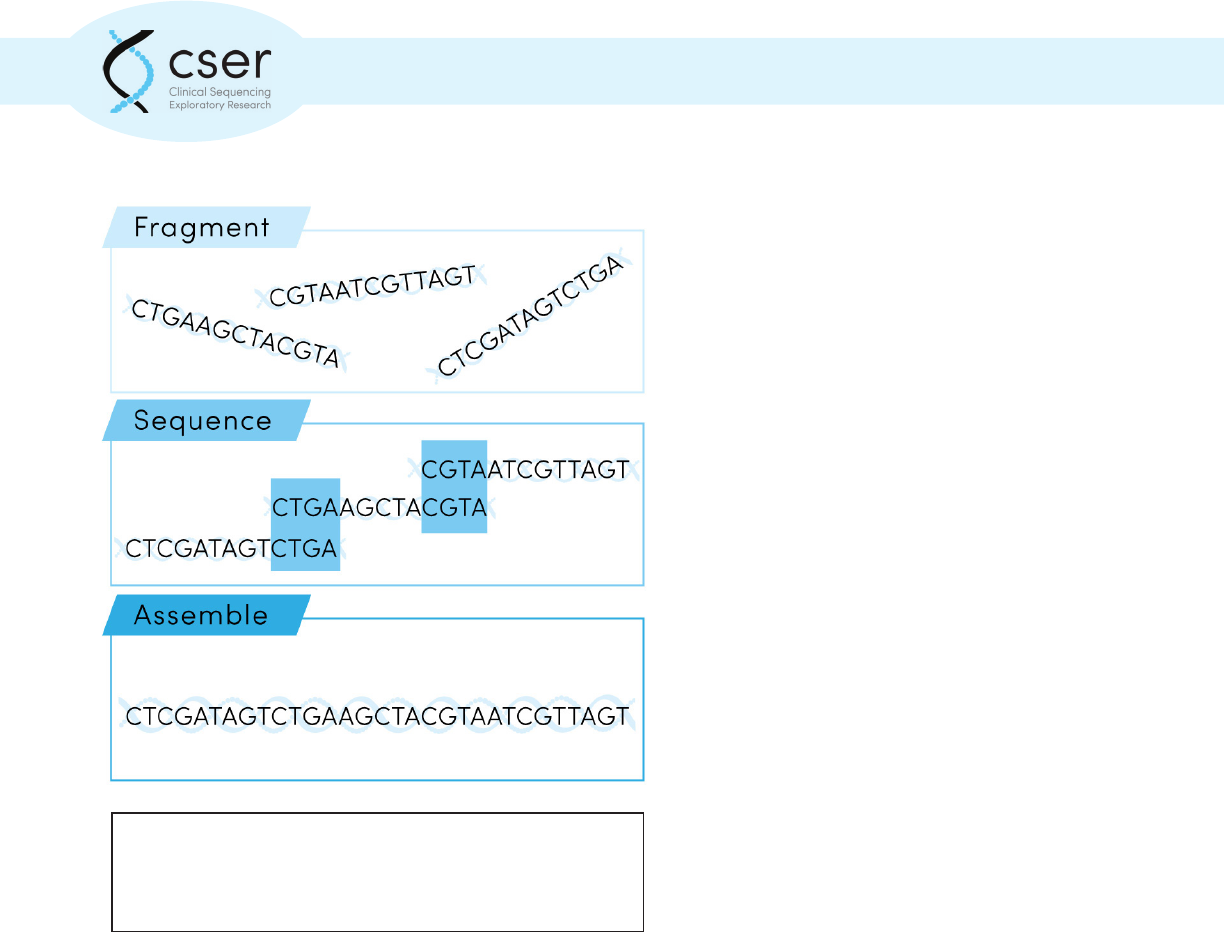
Next Generation Sequencing (NGS), used in diagnostic testing, generally involves determining the
patient’s genetic sequence in millions of short segments, called “reads” (each approximately 100
basepairs in length), assembling the reads into a complete sequence, then determining what genetic
variants are present and interpreting what they mean.
4
1: Introduction and Overview
NGS is extremely exible and has been
implemented for sequencing only a few
genes (e.g., hereditary breast cancer
panels), whole exomes (all of the coding
regions of DNA) or whole genomes (entire
DNA sequence including both coding and
noncoding regions).
NGS is now routinely performed in clinical
diagnostic laboratories that perform
genetic testing and are regulated by CAP
and CLIA certications. However, much is
still unknown about when to use which
kind of test (gene panels, exome testing,
and genome testing), and for what clinical
purpose. There are CPT codes for these
new NGS tests, but insurance coverage is
variable.
For exome or genome sequencing,
potentially millions of variants are
identied that dier between the patient
and the “reference sequence” used for
comparison. Most genetic variants have
little or no known impact on human health, so the variants must be ltered to identify the few that
are medically meaningful. Genomic data from an individual’s parents provides information that can
help lter out benign genetic variants and identify de novo variants (variants that are not inherited).
Generation of the sequence data, variant calling, and variant interpretation are all critical steps for
providing accurate test results.
Genomic testing is often done for an individual patient (singleton) or for a trio (includes patient,
mother, and father); however other formats are possible depending on the disease of interest and
family structure. Not all laboratories handle the testing and interpretation of parental samples in the
same way. In some labs all three individuals are sequenced and interpreted comprehensively at the
same time. In other labs the parental samples are only tested after the patient’s sample.
Next generation sequencing involves determining an
individual’s genetic sequence in millions of short segments
which are then assembled into a complete sequence.
5
1: Introduction and Overview
Making sense of genetic variation
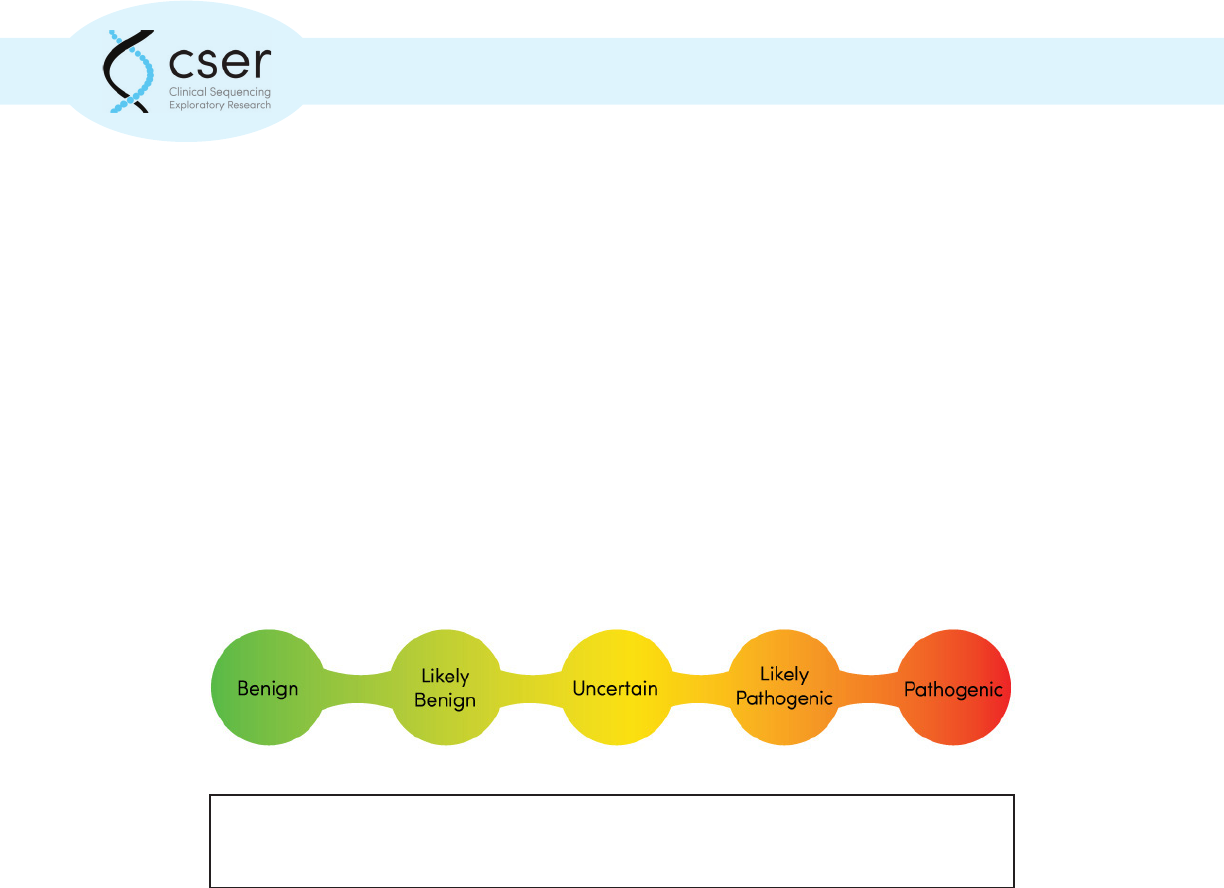
Individual genetic variants are classied by the testing laboratory, to indicate whether the
laboratory believes a variant to be disease causing (pathogenic) and how certain this assessment
is. Laboratories often use a 5-point scale to assign pathogenicity from benign (not disease causing)
to pathogenic, with intervening scores of likely benign, variant of uncertain signicance, and
likely pathogenic. The American College of Medical Genetics and Genomics (ACMG) has published
guidelines for laboratories to use in their interpretation and scoring of genetic variation
2
. However,
laboratories will vary in the types of variants they report. Some laboratories may report only
pathogenic and likely pathogenic variants while others may report variants of uncertain signicance
as well. Laboratories typically do not report benign or likely benign ndings. Sometimes variants
that are associated with causing disease are also called mutations. To reduce confusion, all genetic
changes—whether they cause a medical condition or have no impact at all—are now called variants.
Genomic tests such as exome or genome sequencing can provide dierent kinds of information.
Results that are directly related to explaining a patient’s symptoms or reason for testing are often
called primary ndings, while results that are medically meaningful but unrelated to the reason for
testing are often called secondary or incidental ndings. When using genomic testing to diagnose
patient symptoms, examples of secondary ndings may include genetic risks for future disease,
carrier status (carrying a gene for, but not exhibiting, a condition), and pharmacogenomics ndings
(ndings related to dierences in how a person may process medications). Often laboratories
request information about what kinds of secondary ndings a patient would like reported during the
test ordering and informed consent process. Laboratories vary in how these choices are categorized
and structured.
At this time there is also wide variability among genomic laboratories in the scope and structure of
their result reports. During the development of this toolkit that authors reviewed information from
several dierent labs to help providers make sense of the language they will see in these reports.
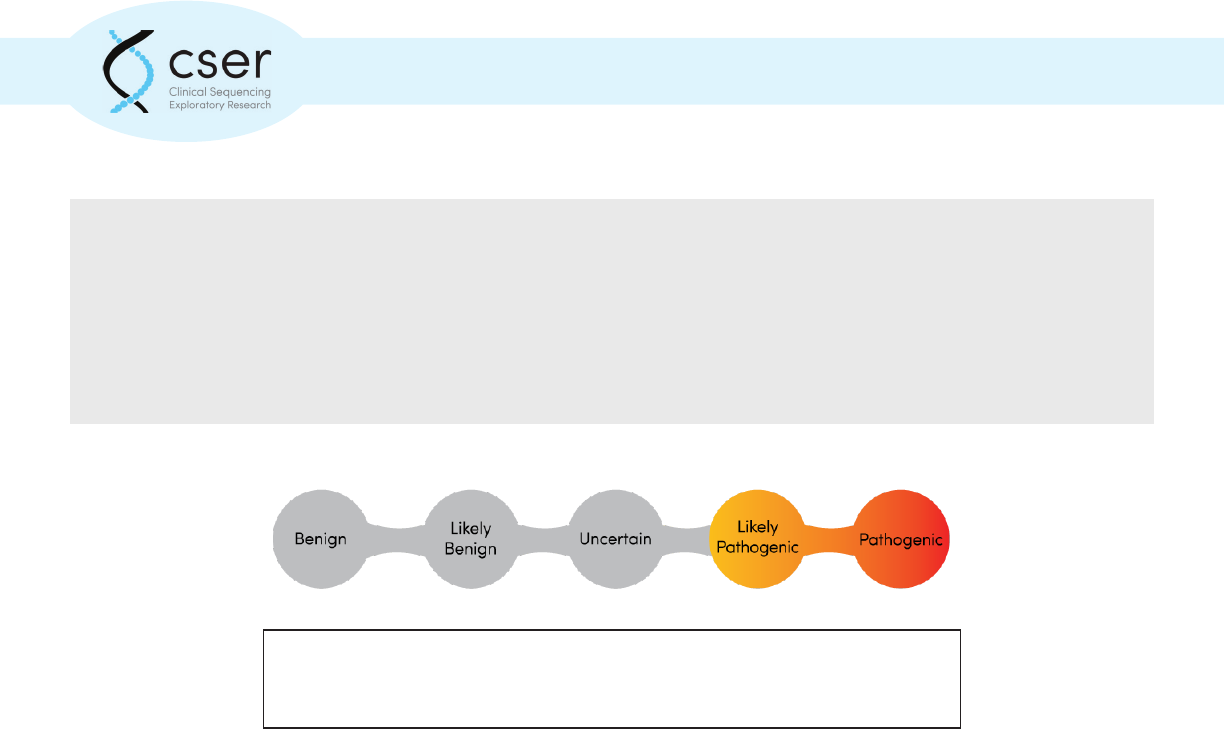
Genomic variants are typically classied on a ve-point scale to indicate the likelihood that the
particular variant is associated with disease.
6
1: Introduction and Overview
Limitations
There are limitations to genomic testing using NGS. A “negative” test report does not exclude the
possibility of an underlying genetic disease. There may also be variants of uncertain signicance
reported that will become better understood over time. Additionally, several types of genetic
variants are not robustly detected by NGS methods. The sensitivity of the test is disease specic and
should be considered before ordering.
It is highly likely that a genetic variant reported is truly present, particularly if the laboratory uses a
second testing method (such as Sanger sequencing) to conrm this. However, it is more dicult to
determine the signicance of a variant. Laboratories do their best to accurately label genetic variants
as pathogenic or likely pathogenic, but a classication may be incorrect. Further, emerging evidence
may lead to reclassication. The original report would then have been a false positive.
Should family members be tested?
Genomic test results may provide information about potential genetic variants and risk factors
among a patient’s family members. When a genetic variant is identied in an individual, it is
important to determine whether relatives are at risk of also having that genetic change and what
follow-up testing might be indicated.
What about genetic discrimination?
Many patients may have questions about who will have access to their test results and how that
information can be used. Genetic discrimination refers to using a person’s genetic information
(test results, family history) against him or her in a harmful way. The Genetic Information
Nondiscrimination Act (GINA), a federal law passed in 2008, largely protects against genetic
discrimination in the areas of health insurance and employment. However, this legislation does not
apply to life, disability, and long-term care insurance. To learn more about GINA, visit http://ginahelp.
org/. Individual states may have additional laws that protect against genetic discrimination.
About this toolkit
This toolkit is organized into dierent sections, based on the dierent categories of results that may
be generated by a genomic test. Each category includes example results and the benets, limitations,
and special considerations associated with each category.
References:
1. Green, R.C., et al. “Clinical Sequencing Exploratory Research Consortium: Accelerating
Evidence-Based Practice of Genomic Medicine.” Am J Hum Genet. 2016 Jun 2;98(6):1051-66.
2. Richards, S. et al. “Standards and Guidelines for Interpretation of Sequence Variants: A joint
consensus recommendation of the American College of Medical Genetics and Genomics and
Association for Molecular Pathology.” Genetics in Medicine. 2015 17: 405-423.

7
1: Introduction and Overview
Resources:
ClinGen (http://clinicalgenome.org): a NIH-funded resource that denes the clinical relevance of
genes and variants for use in medicine and research.
ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/): a public archive of reports about the relationship
between specic genetic variants and associated phenotype (symptoms).
GeneReviews (hp://www.ncbi.nlm.nih.gov/books/NBK1116/): a collecon of chapters, each focused on
an individual genec condion or disease, wrien for healthcare providers by experts in the eld.
GINA (hp://www.ginahelp.org/): an online resource about genec discriminaon and the Genec
Informaon Nondiscriminaon Act.
Naonal Society of Genec Counselors (hp://nsgc.org/): the professional organizaon for genec
counselors, with paent and provider resources and a searchable tool to “nd a genec counselor” near
you.
OMIM (hp://www.ncbi.nlm.nih.gov/omim): an online resource about human genes and associated
phenotype (symptoms).
8
2: Diagnostic Results Related to Patient Symptoms: Pathogenic and Likely Pathogenic Variants
Key Points:
• Pathogenic variants in disease genes related to phenotype (or symptoms) means that a cause
of the patient’s symptoms has been identied.
• Clinically, both pathogenic and likely pathogenic variants are treated the same—as if they are
likely disease causing.
Often when a whole exome or whole genome sequence test is performed, the primary goal is to
answer a diagnostic question about a patient with a specic set of symptoms (phenotype). When a
genetic cause is identied that is believed to account for the symptoms, the result is described as a
primary nding with one or more “pathogenic” or “likely pathogenic” variants in disease genes
related to the phenotype. In other words, there is an answer and a denitive or highly probable
cause to return to the patient and provider.
If the particular variant found has been previously associated with the condition, the variant will
be classied as “pathogenic.” However, frequently, there is insucient evidence that a variant is the
denite cause for symptoms. The term “likely pathogenic” means that the variant most likely has a
harmful eect. When a gene is associated with a disease that overlaps with the patient’s symptoms,
it then represents the likely diagnosis and cause. Sometimes, the gene has been linked to disease
and the clinical features of the condition overlap with the patient’s symptoms, but the exact gene
variant identied in the patient has not been previously observed, making interpretation dicult.
Many factors are considered in assessing whether a variant is pathogenic. Clinically, pathogenic and
likely pathogenic variants are usually treated the same—as if they are likely disease causing—and
clinical management is tailored based on this diagnosis.
Genomic variants are typically classied on a ve-point scale to indicate the
likelihood that the particular variant is associated with disease.
9
2: Diagnostic Results Related to Patient Symptoms: Pathogenic and Likely Pathogenic Variants
Example:
A patient presents with vision loss, obesity, renal failure, and cognitive decits. Genomic
sequencing identies a variant in the BBS10 gene. The BBS10 gene is associated with Bar-
det-Biedl syndrome and is consistent with all of the clinical features in the patient. In addition,
the specic variant has been repeatedly described in the literature in patients diagnosed with
Bardet-Biedl syndrome. Based on this evidence, the variant would be classied as pathogenic.
In contrast, if sequencing identied a dierent variant in BBS10 that had not been previously
observed in either healthy or symptomatic populations, the variant would probably be clas-
sied as “likely pathogenic.” With time and as more individuals are sequenced, many “likely
pathogenic” variants are reclassied to “pathogenic.”
Next Steps to Consider:
• Learn more about the condition with which the patient has been diagnosed through
GeneReviews and other resources
• Referral to genetic services (medical geneticist and/or genetic counselor) for medical
follow-up and discussion of recurrence risks and implications for other family members
• Identication of relevant patient support groups and resources specic to diagnosis
• Identication of relevant research studies or clinical trials specic to diagnosis
Resources
ClinicalTrials.gov (https://clinicaltrials.gov/): a registry and results database of publicly and privately
supported clinical studies of human participants conducted around the world.
GeneReviews (https://www.ncbi.nlm.nih.gov/books/NBK1116/): a resource for clinicians that pro-
vide clinically relevant and medically actionable information for inherited condition. This resource
includes information on diagnostic criteria, management, and information about genetic counseling
for patients and their families. There are chapters available about many, but not all, genetic condi-
tions.
National Society of Genetic Counselors (http://nsgc.org/): the professional organization for genetic
counselors, with patient and provider resources and a searchable tool to “nd a genetic counselor”
near you.
10
3: Uncertain Results Related to Patient Symptoms: Variants of Uncertain Signicance
Key Points:
• Variants of Uncertain signicance have an uncertain relationship to disease.
• It is not recommended that Variants of Uncertain Signicance be used for clinical decision
making.
• International eorts are underway to reclassify VUS variants as benign or pathogenic.
• Finding a VUS is common among large-scale tests like gene panels, whole exome, and whole
genome sequencing.
Sequencing of a gene can identify variants that are dierent from the reference sequence, yet not
well understood. In contrast to genetic variants that have been conrmed to be associated with
increased risk for developing a disease, a Variant of Uncertain Signicance (VUS) has an uncertain
relationship to disease.
There are several reasons why variants are classied as VUS, such as:
a. The eect of the specic genetic alteration on gene function is not known.
b. There are insucient genetic data to denitively conrm that the variant is associated with
risk of developing the disease.
c. The patient is unaected and has no symptoms, or dierent symptoms than those expected
based on the variant found.
As more data accumulate over time, a VUS may be reclassied to likely pathogenic or likely benign.
Until a VUS is reclassied, clinicians are advised not to use a VUS result for clinical decision-making.
Further, the American College of Medical Genetics and Genomics (ACMG) recommends against using
a VUS result for genetic testing in at-risk relatives because the meaning of the result is uncertain
1
.
However, testing of certain relatives can sometimes provide useful data to the testing laboratory to
aid in the eventual reclassication of the variant.
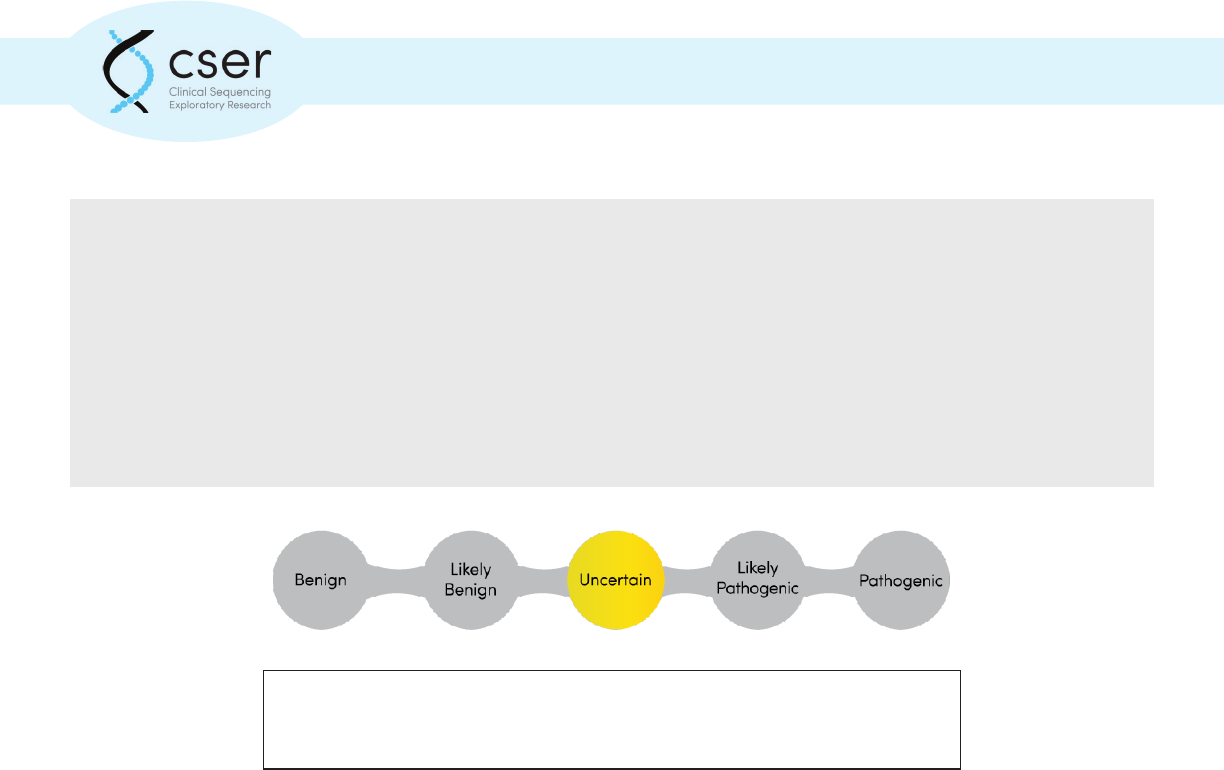
Genomic variants are typically classied on a ve-point scale to indicate the
likelihood that the particular variant is associated with disease.

11
3: Uncertain Results Related to Patient Symptoms: Variants of Uncertain Signicance
International eorts are underway to systematically review all genomic variants, with the objective
of reclassifying VUS results according to best practice guidelines. Many clinical genetic testing
laboratories will, as a matter of policy, issue an amended report when a VUS result is reclassied
to a category considered clinically actionable, and these amended reports are usually sent to the
physician who ordered the test. As reclassication of a VUS may occur years after the original test
was performed, clinicians and patients may consider re-contacting the laboratory that performed
the genetic testing periodically for updates. In addition, resources are available online that enable
clinicians to explore variants identied in their patients. For example, ClinVar (http://www.ncbi.nlm.
nih.gov/clinvar/) is a robust online resource that catalogues the classication of specic genetic
variants, submitted by genetic testing laboratories and experts.
In the case of genetic tests that examine multiple genes, such as multigene panels, whole exome
sequencing, and whole genome sequencing, the likelihood of nding one or more VUS is high; some
studies report VUS in as many as 1 in 3 patients
2
. Individuals undergoing multigene sequencing
tests should receive pre-test education about the fact that such testing increases the likelihood of
a VUS result. In addition, patients of a non-Caucasian ethnic background that has been less well
characterized have an increased likelihood of VUS results.
Example:
A 39-year-old woman diagnosed with breast cancer undergoes gene sequencing for
alterations in the BRCA1 and BRCA2 genes and is found to carry a variant of uncertain
signicance (VUS) in BRCA2. Two years later, the woman is diagnosed with another cancer - a
sarcoma. She undergoes genomic sequencing of both tumor DNA and germline DNA (healthy,
non-tumor DNA). Her germline DNA result shows a pathogenic variant (or mutation) in the
TP53 gene, a well-known tumor suppressor gene associated with Li Fraumeni syndrome.
This new information conrms a diagnosis of Li Fraumeni, a hereditary cancer syndrome
characterized by strikingly increased rates of cancer, in this patient.
Further review of data on the initial BRCA2 variant shows that this variant is now known to be
commonly observed in individuals of Asian ancestry and has been reported in 5 individuals
who were also conrmed to carry pathogenic mutations in other cancer-associated genes. As
a result, the BRCA2 variant is reclassied as “likely benign.”
The initial genetic test identied a VUS in BRCA2. Although BRCA2 is one of the most common
genes implicated in breast cancer, the variant was classied as a VUS because of insucient
information. The fact that a pathogenic germline mutation in TP53 was subsequently
identied in this patient helped conrm that the BRCA2 VUS was likely benign. This woman’s
12
3: Uncertain Results Related to Patient Symptoms: Variants of Uncertain Signicance
family members should be oered testing for the pathogenic TP53 variant to help assess
their cancer risk. Had testing for the BRCA2 VUS been oered to the family members their test
results would have been misleading, since it is the germline TP53 pathogenic variant, not the
BRCA2 VUS, that is responsible for the increased cancer risk in this family.
Next Steps to Consider
• Referral to genetic services (medical geneticist and/or genetic counselor) for medical
evaluation and in-depth discussion of the identied genetic variant
• Contact the testing laboratory periodically for reanalysis of the genetic variant to
determine whether more is known about its disease association
• Explore current knowledge about the specic genetic variant utilizing resources such as
ClinVar
• Identication of relevant research studies specic to the gene or variant identied
• Consider periodic reanalysis of genomic test data as knowledge and databases used in
analysis improves over time
Resources:
ClinicalTrials.gov (https://clinicaltrials.gov/): a registry and results database of publicly and privately
supported clinical studies of human participants conducted around the world.
ClinVar (http://www.ncbi.nlm.nih.gov/clinvar/): a public archive of reports about the relationship be-
tween specic genetic variants and associated phenotype (symptoms).
National Society of Genetic Counselors (http://nsgc.org/): the professional organization for genetic
counselors, with patient and provider resources and a searchable tool to “nd a genetic counselor”
near you.
References:
1. Richards S. et al. Standards and Guidelines for Interpretation of Sequence Variants: A joint
consensus recommendation of the American College of Medical Genetics and Genomics and
Association for Molecular Pathology. Genetics in Medicine (2015); 17: 405-423.
2. Domchek, S. Multiplex Genetic Testing for Cancer Susceptibility: Out on the High Wire without
a net? J Clin. Oncol. 2013; 31: 1267-70.
13
Key Points:
• A negative (normal) result reduces but does not eliminate the possibility that there is a genetic
cause for the patient’s condition.
• Whole genome sequencing may identify genetic causes missed by whole exome sequencing.
• With time, more variants will be recognized as disease causing and reanalysis of whole exome
or whole genome data may be helpful in the future.
With whole exome or whole genome sequencing, when a genetic cause is not identied that is
believed to account for the patient’s symptoms (phenotype), the result is described as “normal” or
“negative” or “none detected” for mutations in disease genes related to phenotype. In other words,
there is no answer and there is no identiable genetic cause for the patient. A normal result reduces
but does not eliminate the possibility that there is a genetic cause for the patient’s condition. Rather,
it indicates that a genetic cause could not be identied.
The limitations of the test should be reviewed with the patient. There are important reasons why a
genetic cause may not be identied by whole exome or whole genome sequencing. Whole exome
sequencing only captures and sequences 1-2% of the genome, and the disease causing variants
could be in non-coding DNA regions that are not targeted for capture or in coding regions that are
not well captured by the test.
On average, more than 95% of most genes are captured with current technology, but coverage
varies by the lab, capture kit, and by gene. The disease causing variant may have been captured and
sequenced, but it may be part of a gene that is not yet associated with disease. The disease causing
variant may have been sequenced and may be present in a known disease causing gene but may
be a novel variant that was not understood to be disease causing. While laboratories can sequence
whole exomes and genomes, there are limitations in the technology and the interpretation of the
data that may allow for a disease-causing genetic change to be missed. Additional limitations include
certain kinds of genetic changes that are not easily detected through sequence based methods,
including repeat expansions and structural chromosome rearrangements. It is important to make
sure any genes of particular interest were adequately interrogated by the chosen test. A negative
genomic test does not rule out a genetic cause for disease.
4: No Findings Related to Patient Symptoms

14
Sequencing a trio, meaning a patient and both of his or her biological parents, can increase the diag-
nostic yield as it allows the laboratory to identify variants that are de novo (not inherited). If only the
aected individual was sequenced, such variants are less likely to be interpreted correctly and less
likely to be reported.
Next Steps to Consider:
• Referral to genetic services (medical geneticist and/or genetic counselor) for medi-
cal evaluation and/or additional testing that may supplement the genomic test
• Consider other types of diagnostic testing for the patient as the cause of symp-
toms or reason for testing has not yet been explained
• Consider periodic reanalysis of genomic test data as knowledge and databases
used in analysis improves over time
Resources
National Society of Genetic Counselors (http://nsgc.org/): the professional organization for genetic
counselors, with patient and provider resources and a searchable tool to “nd a genetic counselor”
near you.
4: No Findings Related to Patient Symptoms
Sequencing an individual as well as both biological parents can increase
diagnosticyield as it allows for the identication of de novo (not
inherited) variation.

15
5: Medically Actionable Secondary Results
Key Points:
• Secondary ndings are additional results that do not directly relate to the reason for testing.
• Patients are often given the opportunity to opt in/out of receiving secondary ndings when a
whole exome or genome sequencing is ordered.
• Common types of secondary ndings include risk of future disease, response to medications,
and carrier status results.
• Many laboratories only report secondary ndings that are medically actionable.
Genomic sequencing is used to aid in the diagnosis of a
patient who, because of symptoms or family history, is
suspected to have an underlying genetic disorder. The term
“secondary ndings” refers to results that do not pertain
to this primary diagnostic question. Secondary ndings are
a diverse category. Some secondary ndings are medically
actionable, meaning they prompt clinical action by the
patient’s medical provider. In the case where genomic
sequencing is being performed to identify future disease
risk in an ostensibly healthy individual, results that are
traditionally thought of as secondary ndings may become
the primary ndings.
The terms “secondary ndings” and “incidental ndings” are often used interchangeably; however,
they are sometimes employed in dierent ways. The secondary ndings label is applied to results
that are unrelated to the diagnostic question, but are nonetheless systematically sought out and
analyzed. Incidental ndings are not sought out, but identied nonetheless. Other terms that have
been used to refer to secondary ndings include “additional ndings,” “o-target results,” and
“unanticipated ndings.”
Secondary ndings, if present, are typically reported for the person undergoing sequencing. When
exome sequencing is completed for relatives of that person, such as parents for trio analysis,
secondary ndings may also be reported in relatives.

16
5: Medically Actionable Secondary Results
Example:
A patient presents with a personal history of peripheral neuropathy. Genomic sequencing
is performed and a pathogenic variant is identied in the MLH1 gene. Pathogenic mutations
in the MLH1 gene are associated with hereditary Lynch syndrome, which increases an
individual’s risk to develop a variety of cancers including colon cancer. However, it is
unrelated to the neuropathy symptoms and would thus not be part of the patient’s diagnostic
result. This result would be classied as a secondary nding, unrelated to symptoms and
reason for testing. Patients with Lynch syndrome are advised to have a colonoscopy with
removal of any polyps every 1-2 years beginning in their mid-20s. This is a highly eective way
to reduce colon cancer risk in individuals with Lynch syndrome. Because the identication of
a pathogenic MLH1 mutation would prompt the patient’s clinician to initiate the colonoscopy
screening protocol at the appropriate age, this is a medically actionable secondary nding.
Genomic testing can identify risk factors for future disease development. Genomic testing can
also provide information about an individual’s carrier status for autosomal recessive diseases.
While carrier status is not expected to impact an individual’s own health or medical care and is not
actionable in the same way as was the secondary nding in the example above, it may be considered
medically actionable by some, particularly by patients of reproductive age and their providers.
Similarly, pharmacogenomics results (results concerning how genes aect a person’s response
to drugs) may be included in secondary ndings. These results would be medically actionable only
if the patient takes a relevant drug. For more information about these types of results, please see
Section 7: Pharmacogenomics Results.
How likely is a medically actionable secondary nding?
Most, if not all, laboratories oering clinical genomic sequencing return medically actionable
secondary ndings of some sort. The likelihood of such a nding depends on how secondary
ndings are dened and analyzed by the laboratory. The American College of Medical Genetics
and Genomics (ACMG) proposed a list of 56 genes with 24 associated phenotypes as a minimum
list of secondary ndings to be systematically analyzed and reported by laboratories oering exome
or genome sequencing
1
. It has been estimated that ~1% of patients undergoing exome sequencing
would receive a secondary nding using this list.
Because it would be inappropriate to initiate medical intervention for an unaected person on the
basis of uncertain information, laboratories typically only report pathogenic or likely pathogenic
variants as secondary ndings
2
. The ancestral background of the patient undergoing sequencing
may inuence the likelihood of a secondary nding. To date, individuals of European Caucasian
ancestry have been overrepresented among cohorts undergoing genetic sequencing. Thus, there

17
5: Medically Actionable Secondary Results
may be more evidence available to assess pathogenic and likely pathogenic variants in this group.
Causative variants identied in non-European Caucasian populations such as African or Asian may
go unrecognized due to lack of data, may be labeled variants of uncertain signicance (VUS), or
may not be reported.
Depending on a specic laboratory’s policy, there is often an opportunity for patients to opt in or out
of receiving such secondary ndings.
What if there are no medically actionable secondary ndings?
Most patients undergoing genomic sequencing will not have a medically actionable secondary
nding. This means there were no genetic variants identied that suggested a high likelihood of an
actionable genetic condition that was unanticipated due to clinical signs and symptoms. Laboratories
set a high bar for returning secondary nding results. Therefore, a lack of medically actionable
secondary ndings is not a clean bill of health, and does not reduce a person’s risk for future health
problems such as cancer, cardiac arrhythmia, or vascular disease, since in the general population,
most cases of such health problems are not attributable to the types of rare genetic conditions
revealed as secondary ndings from genetic sequencing. In addition, false negative results are
possible due to a number of technological and interpretive limitations. Secondary ndings must be
examined in the context of the individual’s medical history and family history.
Why might a lab report secondary ndings that are not medically actionable?
Many laboratories only report secondary ndings that are not medically actionable if the patient
actively chooses to receive such information. However, some laboratories may report on secondary
ndings that are not medically actionable but may be highly predictive. For example, pathogenic
mutations in the SOD1 gene are associated with familial amyotrophic lateral sclerosis (ALS), also
known as Lou Gehrig’s disease. People with a pathogenic mutation in this gene have a 90% chance
to develop ALS by the time they are in their 70s. This nding would not be considered medically
actionable because there are currently no therapies to prevent or delay ALS, or slow its progress.
Despite this, a result of this sort may be desired. Patients electing to learn this type of information
should consider possible benets (such as enhanced ability to plan) with possible negative
consequences (such as the risk of life insurance discrimination, or increased anxiety).
Age Matters
Most laboratories that return medically actionable secondary ndings routinely do so regardless of
the age of the patient. Laboratories that oer non-medically actionable secondary ndings generally
oer this only for adults who are competent to provide informed consent to receive this category of
ndings.
18
5: Medically Actionable Secondary Results
References:
1. Green, R. C., et al. “ACMG Recommendations for Reporting Incidental Findings in Clinical
Exome and Genome Sequencing.” Genet Med. 2013 Apr;15(7):565-574. https://www.acmg.
net/docs/ACMG_Releases_Highly-Anticipated_Recommendations_on_Incidental_Findings_in_
Clinical_Exome_and_Genome_Sequencing.pdf
2. Amendola, L.M., et al. “Actionable exomic incidental ndings in 6503 participants: challenges
of variant classication.” Genome Res. 2015 Mar;25(3):305-15.
Resources:
Anticipate and Communicate: Ethical Management of Incidental and Secondary Findings in the
Clinical, Research, and Direct-to-Consumer Contexts, from the Presidential Commission for the Study
of Bioethical Issues http://bioethics.gov/sites/default/les/FINALAnticipateCommunicate_PCSBI_0.pdf
19
6: Common Risk Allele Results
Key Points:
• Common complex disorders such as heart disease, diabetes, and most cancers develop as a
result of both genetic and environmental factors.
• The association of a common risk allele with disease is often modest, as is its impact on
clinical care.
• Interpretation of common risk alleles for clinical management can be challenging.
Dierences in the DNA sequence at a specic location
in a gene are called variants, or alleles. Alleles with a
high population frequency, typically dened as >1%, are
referred to as polymorphisms. These small dierences
in DNA sequence, or genetic variation, may or may not
aect gene function. However, some common variants
can interfere with a biological process, leading to illness,
typically in combination with other factors. Such conditions
are considered to have a genetic basis, and are typically
classied as “common complex” disorders.
In contrast to Mendelian disorders (e.g. Huntington’s
disease, sickle cell anemia) in which variation in a single
gene causes disease, common complex disorders, such
as heart disease, diabetes, and most cancers, develop
as a result of both genetic and environmental factors.
Because common complex diseases can be associated
with alterations in many dierent genes, and because
each of these alterations is usually associated with only
small increases in risk, the nding of a common risk allele
has much less impact on clinical care than nding a gene
mutation associated with a Mendelian disorder.
Risk alleles for common complex diseases are usually dened by the minor, or least common, allele
frequency (MAF). This allows for dierentiation between common and rare alleles in the population.
The MAF of common risk alleles can range from 5% to 50%. However, just because an allele is
common does not necessarily mean it has a meaningful impact on disease susceptibility.
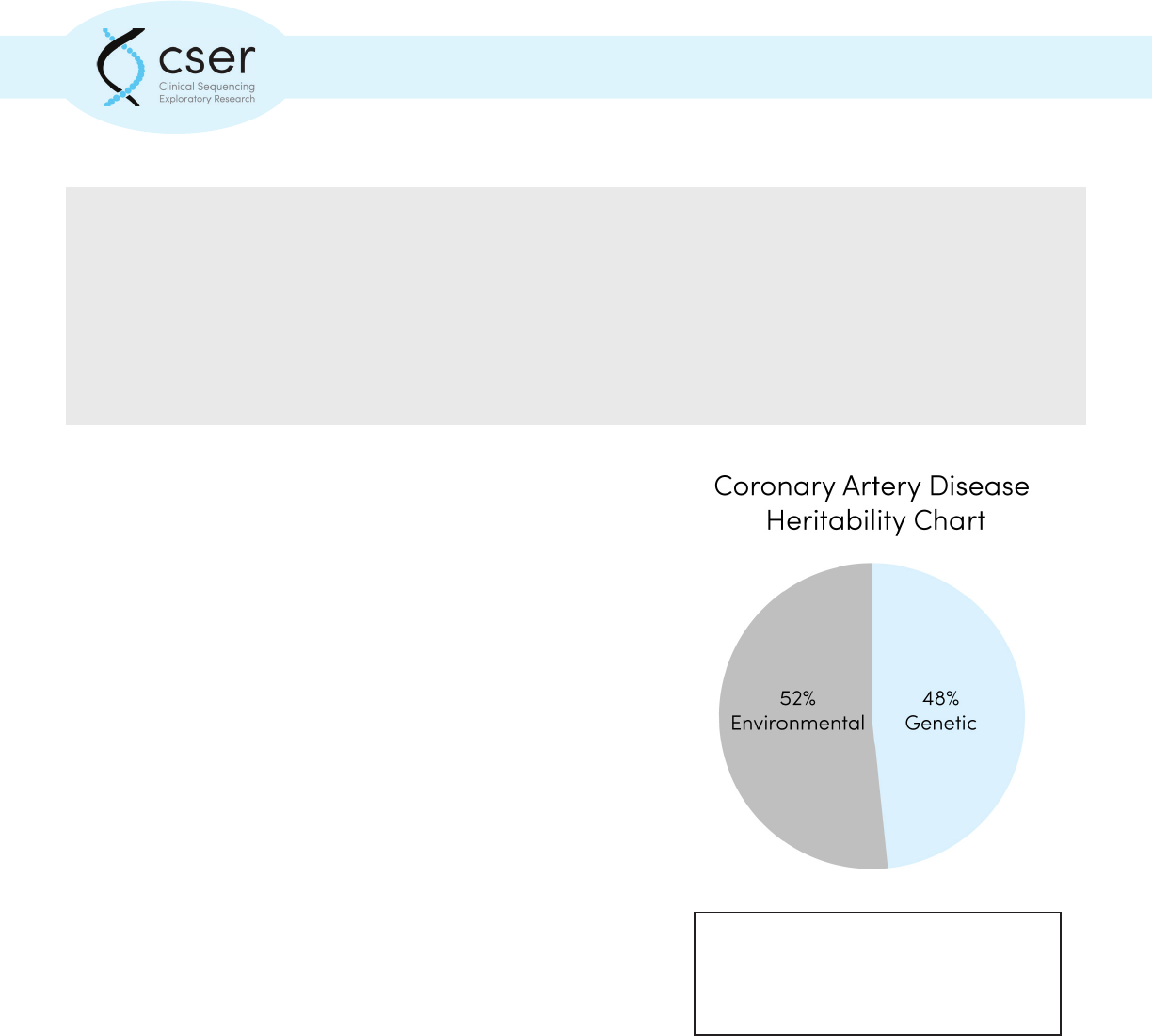
Common complex disorders, such as
coronary artery disease, develop as a result
of multiple genetic and environmental
factors.
20
6: Common Risk Allele Results
Common risk alleles are often detected by genome-wide association studies (GWAS)
1,2
. GWAS
are a type of case-control study in which people with the condition being studied are compared to
similar people without the condition. Each person’s complete set of DNA, or genome, is surveyed
by examining a strategically selected “panel” of genetic markers that tag areas of known variation,
called single nucleotide polymorphisms (SNPs).
If certain genetic variations are found to be more frequent in people with the disease compared to
people without disease, the variant alleles are said to be “associated” with the disease. The presence
of an association suggests that the variant, or some other nearby variant, is inuencing disease
susceptibility.
The association of an allele with disease is a measure of statistical, not clinical signicance. Eects
are often modest, and the associated variants themselves may not directly cause the disease.
Given this, genetic tests for such conditions can only provide an estimate of probability or risk. For
common diseases, the presence of a high-risk allele may only mildly increase the chance of disease.
Further, there are currently no validated ways of combining multiple risk alleles for the same
disease. These limitations can make the interpretation of common risk alleles challenging.
Examples:
One risk allele that is relatively common in the population and that has been associated
with an increased risk for disease susceptibility is factor V Leiden and risk for deep venous
thrombosis (DVT). Between 3- 8% of people of European descent carry one copy of the
factor V Leiden allele and 1 in 5,000 people have two copies. Risk of DVT for individuals in
the general population is 1 in 1,000. However, having one copy of the factor V Leiden allele
increases the risk of DVT from 1 in 1,000 to 3-8 in 1,000. Having two copies raises the risk to
as high as 80 in 1,000. A test that identies the factor V Leiden allele can have implications for
clinical management and may indicate the need for preventative measures to reduce clotting
risk
2
.
In contrast, variants in the MTHFR gene have been associated with increased risk of neural
tube defects and cardiovascular disease; however, 60-70% of individuals in the general
population have one of the two most common MTHFR gene polymorphisms. Most of these
individuals do not develop disease. Therefore, a genetic test that identies one of these
MTHFR gene variants has no real clinical implications.
21
6: Common Risk Allele Results
In the case of breast cancer, several alleles that
increase susceptibility have been identied.
Pathogenic mutations in the BRCA1 and
BRCA2 genes associated with the Mendelian
disorder Hereditary Breast and Ovarian Cancer
Syndrome are associated with lifetime risks for
breast cancer of 40-80%. In contrast, more than
70 other common alleles have been associated
with breast cancer susceptibility, most of which
confer only a mild to moderate increase in
risk. Thus, identifying one of these common
alleles would not have the same implications
for medical management as would nding
a pathogenic mutation in BRCA1 or BRCA2.
To what extent the associated symptoms
are expressed in the presence of the variant
is captured by a term called penetrance. If
it is not always expressed, it is considered
incompletely penetrant.
If an associated allele is identied, it may explain disease susceptibility. Common risk alleles with
a known association with a condition can inform an individual of an increased or decreased risk
of developing the condition in question; however, the degree of certainty is often unknown. The
presence of a common risk allele can indicate a need for increased surveillance, while a negative
result implies a risk similar to the general population.
Common risk alleles have unclear implications for family members. In addition, the clinical
sensitivity of tests for common risk alleles is not necessarily high. Common complex diseases are
caused by multiple genetic and environmental factors, many of which remain unknown.
References:
1. Hirschhorn, J.N., Lohmueller, K., Byrne, E., Hirschhorn, K. “A comprehensive review of genetic
association studies.” Genet Med. 2002 Mar-Apr;4(2):45-61.
2. Raychaudhuri, S. “Mapping rare and common causal alleles for complex human diseases.”
Cell. 2011 Sep 30;147(1):57-69.
3. Geerts, W.H., et al. “Prevention of Venous Thromboembolism: American College of Chest
Physicians Evidence-Based Clinical Practice Guidelines (8
th
Edition).” Chest. 2008;133(6_
suppl):381S-453S.
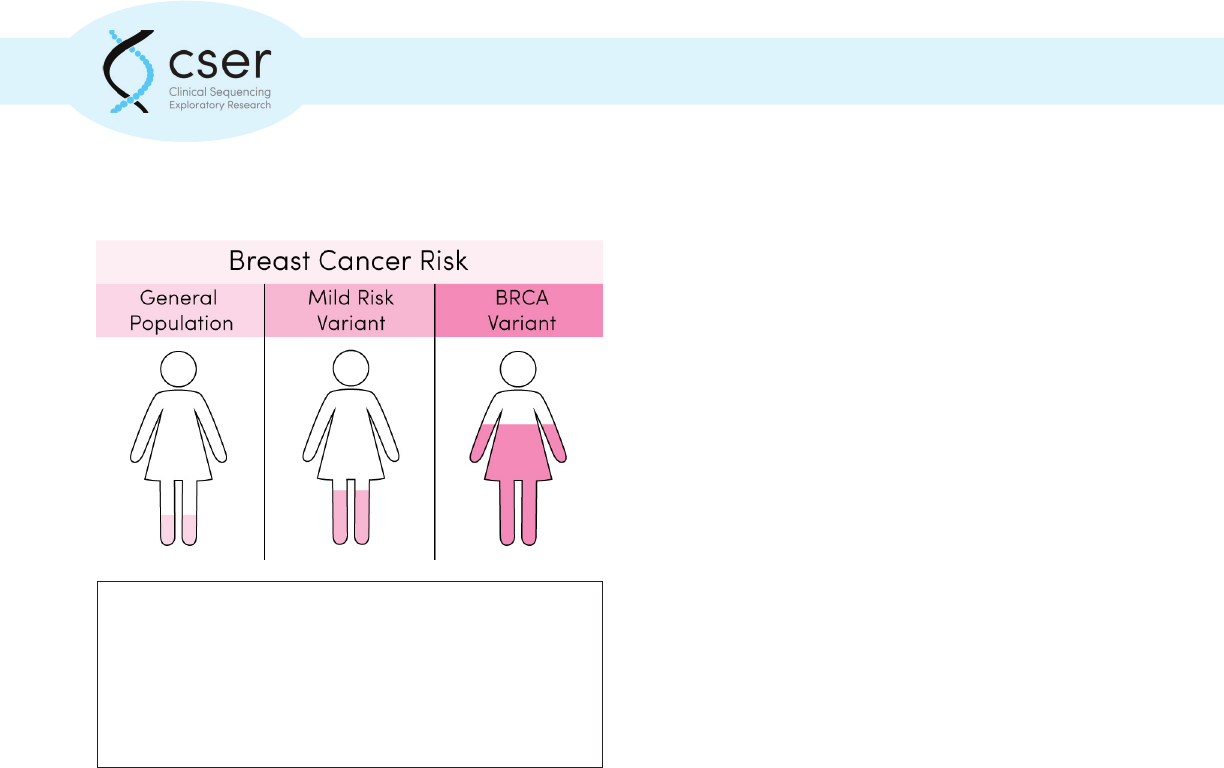
The average woman in the United States has about a 1
in 8, or 12% chance of developing breast cancer during
her lifetime. Genetic variants in highly penetrant genes
(BRCA1 and BRCA2) confer a signicantly higher risk of
being diagnosed with breast cancer, while changes in other
genes have a more moderate impact on risk.
22
Next Steps to Consider
• Use of risk allele information to guide medical management is rarely done in the
absence of a practice guideline
• Explore current knowledge about the specic genetic variant utilizing resources
such as the medical literature, professional guidelines and NHGRI GWAS catalog
• In most cases, testing of family members is not recommended apart from a
specic practice guideline
Resources:
dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/:) a public-domain archive for a broad collection
of simple genetic polymorphisms.
Human Genome Variation (HGV) database (http://www.nature.com/hgv/): a searchable online
database of genome variations published in a variety of peer-reviewed sources. HGV is searchable
and able to be ltered by dierent variables, including specic disease, gene, population or region.
Iles, M.M. 2008. “What can genome-wide association studies tell us about the genetics of common
disease?” PLoS Genet. 2008 Feb;4(2):e33.
OMIM (http://www.ncbi.nlm.nih.gov/omim): an online resource about human genes and associated
phenotype (symptoms).
Vineis P., Schulte, P., McMichael, A.J.. “Misconceptions about the use of genetic tests in populations.”
Lancet. 2001 Mar 3;357(9257):709-12.
References
1. Hirschhorn, J.N., Lohmueller, K., Byrne, E., Hirschhorn, K. “A comprehensive review of ge-
netic association studies.” Genet Med. 2002 Mar-Apr;4(2):45-61.
2. Raychaudhuri, S. “Mapping rare and common causal alleles for complex human diseases.”
Cell. 2011 Sep 30;147(1):57-69.
3. Geerts, W.H., et al. “Prevention of Venous Thromboembolism: American College of Chest
Physicians Evidence-Based Clinical Practice Guidelines (8th Edition).” Chest. 2008;133(6_
suppl):381S-453S.
6: Common Risk Allele Results
23
7: Carrier Status Results
Key Points:
• A carrier is usually asymptomatic due to having one working and one non-working copy of a
gene.
• There is typically a risk for an aected child when both parents are carriers of the same
genetic variant.
• Carrier status can be discovered in various clinical testing scenarios; testing may be ordered
specically looking for carrier status for reproductive planning or to diagnose a symptomatic
individual.
A carrier is an individual who has one working and one non-working copy of a gene. A carrier is
capable of passing on a genetic variant associated with recessive disease (autosomal recessive or
x-linked recessive) to their ospring. A carrier usually does not display disease symptoms associat-
ed with that variant, although in some rare cases a carrier might exhibit some symptoms.
In an individual with a recessive disease, both copies of the gene have variants associated with dis-
ease. Autosomal recessive diseases are typically not seen in every generation of an aected family
and are equally likely to occur in males and females. One example of an autosomal recessive dis-
ease is cystic brosis. Both parents of an individual with an autosomal recessive disease likely carry
one copy of the altered gene.
Females have two X chromosomes, and males have one X and one Y chromosome. For X-linked
conditions, women are typically carriers and have fewer if any symptoms, while males are aected.
Males are at an increased risk of disease because they only have one copy of the X chromosome and
therefore only one copy of the many genes located on the X chromosome. If one of these genes is
not working, there is not another copy to compensate.
A female carrier for an X-linked recessive disease has a 50% chance of passing on the variant in each
pregnancy. Sons that inherit the variant would be expected to be aected with the condition, while
daughters with the variant are less likely to exhibit symptoms. An example of an X-linked reces-
sive disease is Hemophilia A. A male with an X-linked condition will pass on the variant to each of
his daughters (because the variant is on his X chromosome, which he passes on to each female child)
and none of his sons (because each son receives only a Y chromosome from his father).
Carrier status can be discovered in a variety of clinical testing scenarios. Tests may be ordered spe-
cically looking for carrier status, identifying people who carry one copy of a gene variant that can be
24
7: Carrier Status Results
passed on to a child. This type of testing is currently oered to individuals who have a family history
of a genetic disease, people in certain ethnic groups with an increased risk of specic genetic dis-
eases (population screening), and people concerned about their risk of having a child with a genetic
disease.
All individuals are carriers of multiple genetic diseases. However, rarely are partners having a child
both carriers for the same autosomal recessive condition. If couples undergo carrier screening, the
test results can provide information about their risk of having a child with a genetic disease. In this
scenario the carrier status would be considered a primary result, as it is related to the reason the
test was ordered. Carrier testing can be ordered in various ways, including targeted single gene dis-
ease testing, panel testing of multiple genetic diseases, or less commonly through a broad genomic
test looking for genetic variants throughout the genome or exome.
Carrier status information can be used for reproductive planning and may provide information to
other relatives about a possible shared genetic variant. In rare cases when carriers can exhibit symp-
toms, a carrier result could inform medical management for the individual being tested.
Example:
A woman has preconception genome sequencing to plan for a future pregnancy. She is found
to be a carrier of a pathogenic mutation for cystic brosis. Her husband then has genetic
testing to see if he is a carrier of the same mutation. He is negative, meaning a cystic brosis
associated variant was not identied. Based on these results, this couple are very unlikely to
have a child with the condition. However children will have a 50% chance of being an unaf-
fected carrier like their mother.
Carrier status can also be revealed as a secondary nding, or incidental nding when the primary
testing indication is for another reason (e.g., diagnosis of a symptomatic individual) that is unrelat-
ed to assessing carrier status. In this case the carrier result may be relevant to the patient, siblings,
parents, or other relatives for future reproductive planning. As above, in some rare cases in which
carriers exhibit symptoms, a carrier result could inform medical management.
Limitations
There are some important limitations to be aware of when interpreting carrier results from a ge-
nomic test. Carrier testing is a screening test. If one has a negative carrier test result, there is still
a residual risk of being a carrier due to the possibility of a genetic variant that was not detected or
reported. Sequencing does not identify all types of variants. This limitation means that the most
25
7: Carrier Status Results
common variants for some well-known and commonly tested for diseases such as spinal muscular
atrophy (SMA) and Fragile X are cannot reliably be detected by NGS. In addition, many laboratories
do not routinely report variants of uncertain signicance (VUS) related to carrier status results.
An individual may still be a carrier of (or aected with) a disease if no variants or only one variant is
found in the relevant gene.
Some variants (and associated genetic diseases) are more common in certain ethnic groups, due to
a single genetic variant or set of common genetic variants within that population. However, variants
in all genes can occur in any population. It is important to note whether a test interrogates a set of
common variants, or sequences entire genes (and identies both common and rare variants). This,
as well as the carrier frequency within the patient’s specic population group, is important when
determining risk related to a negative result.
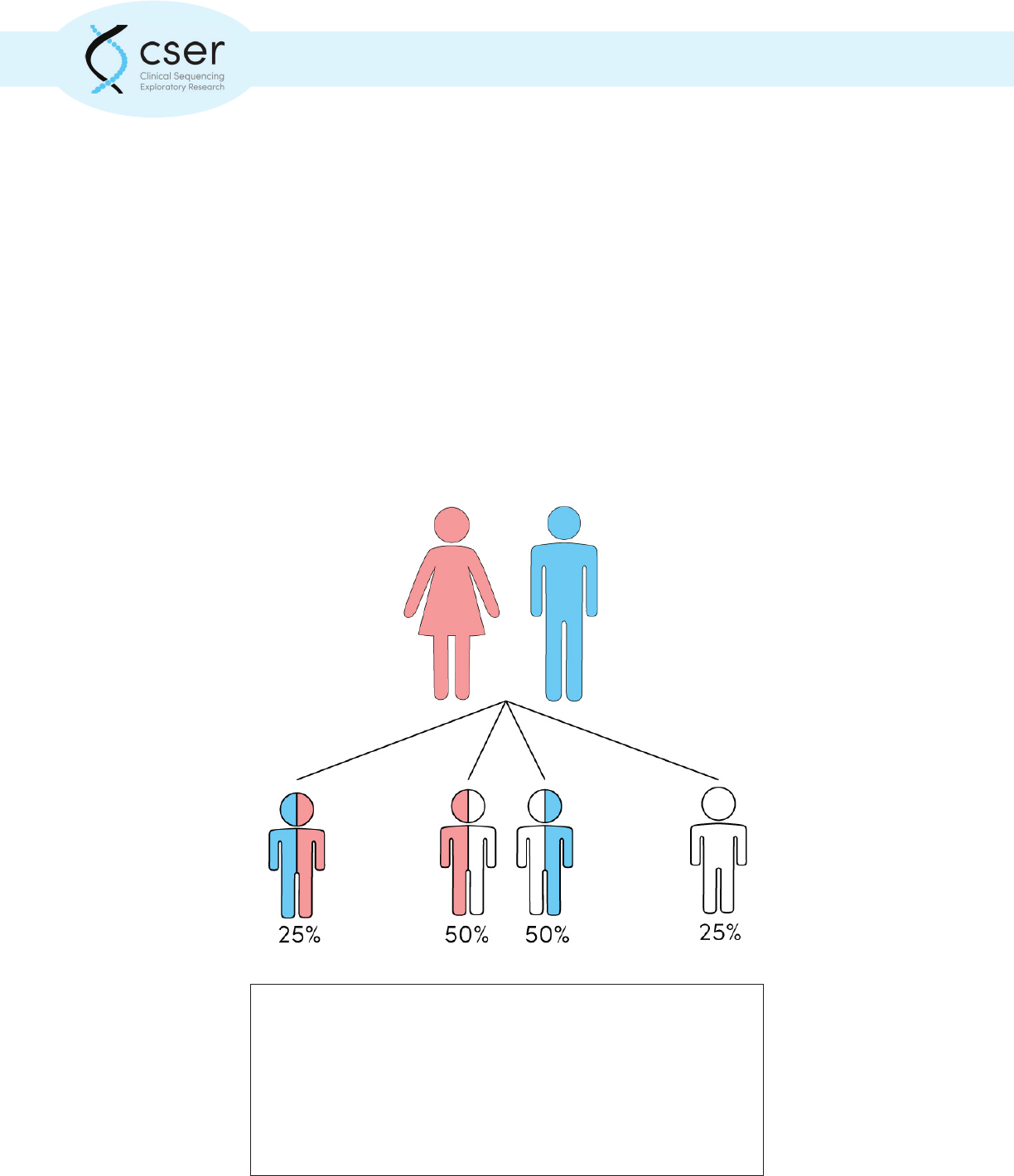
A carrier for a recessive condition is asymptomatic and has a pathogenic
genetic variant in just one of their two copies of the associated gene.
Parents that are both carriers of the same recessive condition have a 25%
chance that a child will inherit the pathogenic variant from both parents
and be aected with the condition. There is a 50% chance that the child
will inherit a single pathogenic variant from one parent and will also be
an asymptomatic carrier. There is a 25% chance that the child will inherit
neither variant and will not be aected or a carrier.
26
Next Steps to Consider:
• Learn more about the recessive condion with which the paent has been diagnosed through
GeneReviews, OMIM and other resources
• Referral to genec services (medical genecist and/or genec counselor) for in-depth
discussion of reproducve risks and implicaons for family members
• Carrier tesng of paent’s partner for the specic gene/genec condion idened to further
clarify risk of having a child with the recessive disorder
Resources:
GeneReviews (https://www.ncbi.nlm.nih.gov/books/NBK1116/): a resource for clinicians that provide
clinically relevant and medically actionable information for inherited condition. This resource
includes information on diagnostic criteria, management, and information about genetic counseling
for patients and their families. There are chapters available about many, but not all, genetic
conditions.
National Society of Genetic Counselors (http://nsgc.org/): the professional organization for genetic
counselors, with patient and provider resources and a searchable tool to “nd a genetic counselor”
near you.
7: Carrier Status Results

27
8: Pharmacogenomic Results
Key Points:
• Pharmacogenetic information can play an important role in identifying individuals at risk for
reduced therapeutic response or at risk for toxicity when given normal doses of particular
medications.
• A comprehensive table of medications and their pharmacogenomic biomarker labeling
information is available for FDA approved medications.
• Gene-drug guidelines to help prescribers with drug selecon and dosing can be found at hps://
cpicpgx.org/
Pharmacogenetic results
from exome or whole
genome sequencing refer to
genetic variants associated
with dierential responses to
medications. These genetic
variants may result in
variable rates of medication
clearance or metabolism.
Certain variants may
increase a patient’s immune
response to a medication.
Pharmacogenetics results can
alert providers to a patient’s
risk for reduced or absent
therapeutic response,
or to possible toxicity-
related adverse events.
These can be prevented
with medication choice,
dose adjustments, or both.
Variation within the CYP2C9 gene alters metabolism of many commonly used medications including
warfarin and valproic acid. HLA gene variants may cause a patient to have severe adverse reactions
when taking certain medications such as carbamazepine. To learn more about specic gene-drug
interactions visit PharmGKB (https://www.pharmgkb.org/).
Genomic variation can alter an individual’s response to medication. Phrarmacogenetic
test results may indicate a need to alter dosage or selection of an alternative
therapeutic.
28
8: Pharmacogenomic Results
Example:
The American College of Medical Genetics and Genomics (ACMG) has proposed a list of
56 genes which should be explored as part of secondary analysis when clinical exome or
genome sequencing is conducted. One is the RYR1 gene. Some pathogenic variants in RYR1
have been shown to increase susceptibility to malignant hyperthermia when halothane
gases, used as a general anesthetic, or succinylcholine, a short-acting muscle relaxant, are
administered. Malignant hyperthermia can cause a fast and potentially fatal rise in body
temperature and severe muscle contractions. Individuals known to have an increased
susceptibility to this reaction because of pathogenic variants in RYR1 should be given
alternative medications when undergoing general anesthesia.
Many researchers are studying the return of pharmacogenetic results to the patient’s Electronic
Medical Record (EMR) when exome or whole genome sequencing is used. Therefore, clinical
sequence data may be analyzed in the relatively near future for important drug response variants
associated with professional guidelines or recommendations.
Using pharmacogenomics information to inform medication selection, dosing or both may reduce
risk for adverse drug reactions. Consideration of a patient’s other current medications or medical
conditions that inhibit or induce genetically normal or dierential processes are essential, in
addition to consideration of dierential drug response due to genetic variants. A comprehensive
table of medications and their pharmacogenomic labeling information is available for FDA approved
medications:
http://www.fda.gov/drugs/scienceresearch/researchareas/pharmacogenetics/ucm083378.htm
Next Steps to Consider
• Consider currently prescribed medications in light of pharmacogenomic results
• Consult pharmacogenomic results as new medications are being considered and pre-
scribed
• Explore current knowledge about the specic genetic variant(s) utilizing resources such as
CPIC and PharmGKB
Resources:
PharmGKB (https://www.pharmgkb.org) - PharmGKB is a NIH funded pharmacogenomics resource
developed by Stanford University that seeks to aid researchers in understanding how genetic
variation contributes to dierences in drug reactions.
Clinical Pharmacogenetics Implementation Consortium (CPIC) (http://cpicpgx.org/) - CPIC creates and
publishes peer-reviewed guidelines for the implementation of pharmacogenetic information into
medical practice.

29
9: Looking Forward
While a patient’s DNA sequence will not change over time, our understanding of his or her sequence
will change, especially given our improving knowledge of genomics and disease. With time, more
variants will be recognized as disease causing and our understanding of currently identied variants
may change. Reanalysis of patient exome or whole genome data may be appropriate in the future.
This toolkit and its resources are meant to provide an introduction to genomic testing for non-genetics
healthcare providers. While this guide is certainly not “the nal word” on these kinds of test results,
as the eld is changing so rapidly, we believe it will provide useful guidance in understanding the
results of today’s tests.

30
Glossary
Autosomal recessive: genetic conditions that occur only when mutations are present in both copies
of a given gene (i.e., the person is homozygous for a mutation, or carries two dierent mutations of
the same gene, a state referred to as compound heterozygosity).
[Source – NCI Dictionary of Genetics Terms]
Benign (variant): an alteration in a gene distinct from the normal, wild-type allele that does.
[Source – Illustrated Glossary]
Carrier frequency: the proportion of individuals in a population who have a single copy of a specic
recessive gene mutation; also sometimes applied to the prevalence of mutations in dominantly
acting genes such as BRCA1 and BRCA2. Also called carrier rate.
[Source – NCI Dictionary of Genetics Terms]
Carrier: an individual who has a recessive, disease-causing variant at a particular location on one
chromosome of a pair and a typically functioning allele at that location on the other chromosome.
[Source – CSER Consortium Practitioner Education Working Group]
Clinical sensitivity: the frequency with which a test yields a true positive result among individuals
who actually have the disease or the gene mutation in question. A test with high sensitivity has a low
false-negative rate and thus does a good job of correctly identifying aected individuals.
[Source – NCI Dictionary of Genetics Terms]
De novo (variant): an alteration in a gene that is present for the rst time in one family member as
a result of a mutation in a germ cell (egg or sperm) of one of the parents, or a mutation that arises in
the fertilized egg itself during early embryogenesis. Also called a new mutation.
[Source – NCI Dictionary of Genetics Terms]
Exome: the exome is the small subset (1-2%) of an individual’s entire genetic sequence, or genome,
that directly instructs the building of a particular protein. Although a small fraction of the genome,
the exome includes the sequence of approximately ~22,000 genes.
[Source – CSER Consortium Practitioner Education Working Group]
False negative: a test result which indicates that an individual is unaected and/or does not have
a particular gene mutation (variant) when he or she is actually aected and/or does have a gene
mutation (variant); i.e., a negative test result in an aected individual.
[Source – Illustrated Glossary]

31
False positive: a test result which indicates that an individual is aected and/or has a certain gene
mutation (variant) when he or she is actually unaected and/or does not have the mutation (variant);
i.e., a positive test result in a truly unaected individual.
[Source – Illustrated Glossary]
Genetic variant: a change in the DNA sequence as compared to a reference sequence that may
or may not have an impact on protein function or disease state. Terms such as mutation and
polymorphism have been largely replaced by ‘variant’ to simplify terminology.
[Source – CSER Consortium Practitioner Education Working Group]
Genome: the entire set of genetic instructions found in a cell. In humans, the genome consists
of 23 pairs of chromosomes, found in the nucleus, as well as a small chromosome found in the
cells’ mitochondria. Each set of 23 chromosomes contains approximately 3.1 billion bases of DNA
sequence.
[Source – Talking Glossary of Genetic Terms]
Genome-wide association studies: a genome-wide association study (GWAS) is an approach used
in genetics research to associate specic genetic variations with particular diseases. The method
involves scanning the genomes from many dierent people and looking for genetic markers that can
be used to predict the presence of a disease.
[Source – Talking Glossary of Genetic Terms]
Likely pathogenic variant: a DNA change that is most likely causing a deleterious eect on an
encoded protein product that accounts for the observed symptoms.
[Source – CSER Consortium Practitioner Education Working Group]
Medically actionable: used to alter the treatment or surveillance of a patient.
[Source – CSER Consortium Practitioner Education Working Group]
Next generation sequencing: a high-throughput method used to determine a portion of the
nucleotide sequence of an individual’s genome. This technique utilizes DNA sequencing technologies
that are capable of processing multiple DNA sequences in parallel. Also called massively parallel
sequencing and NGS.
[Source – NCI Dictionary of Genetics Terms]
Glossary

32
Novel variant: a newly discovered, distinct genetic alteration; NOT the same as new or de novo
variant (or mutation). Also called novel mutation.
[Source – NCI Dictionary of Genetics Terms]
Pathogenic: a genetic alteration that increases an individual’s susceptibility or predisposition
to a certain disease or disorder. When such a variant (or mutation) is inherited, development of
symptoms is more likely, but not certain. Also called deleterious mutation, disease-causing mutation,
predisposing mutation, and susceptibility gene.
[Source – NCI Dictionary of Genetics Terms]
Penetrance: a characteristic of a genotype; it refers to the likelihood that a clinical condition will
occur when a particular genotype is present.
[Source – NCI Dictionary of Genetics Terms]
Pharmacogenomics: a branch of pharmacology concerned with using DNA and amino
acid sequence data to inform drug development and testing. An important application of
pharmacogenomics is correlating individual genetic variation with drug responses.
[Source – Talking Glossary of Genetic Terms]
Phenotype: the observable characteristics in an individual resulting from the expression of genes;
the clinical presentation of an individual with a particular genotype.
[Source – NCI Dictionary of Genetics Terms]
Polymorphism: a common mutation. “Common” is typically dened as an allele frequency of at
least 1%. All genes occur in pairs, except when x and y chromosomes are paired in males; thus a
polymorphism with an allele frequency of 1% would be found in about 2% of the population, with
most carriers having one copy of the polymorphism and one copy of the normal allele.
[Source – NCI Dictionary of Genetics Terms]
Primary result: alterations in a gene or genes that are relevant to the diagnostic indication for
which the test was ordered.
[Source – CSER Consortium Practitioner Education Working Group]
Reference sequence: the ‘standard’ sequence of DNA for a particular organism that is used to
compare new sequence data against for alignment and identication of variation.
[Source – CSER Consortium Practitioner Education Working Group]
Glossary

33
Repeat expansion: Some areas within the human genome contain small repetitive sequences of
DNA (i.e. CAG). The number of repeats at a particular genomic location is typically stable. However,
an increase in repeat number has been associated with several human diseases such as Huntington
Disease and Fragile X syndrome.
[Source – CSER Consortium Practitioner Education Working Group]
Sanger sequencing: a low-throughput method used to determine a portion of the nucleotide
sequence of an individual’s genome. This technique uses polymerase chain reaction (PCR)
amplication of genetic regions of interest followed by sequencing of PCR products.
[Source – NCI Dictionary of Genetics Terms]
Secondary result: refers to genomic test results that do not pertain to the primary diagnostic
question or reason for testing. Also referred to as an incidental nding.
[Source – CSER Consortium Practitioner Education Working Group]
Single nucleotide polymorphism: DNA sequence variation that occurs when a single nucleotide
(adenine, thymine, cytosine, or guanine) in the genome sequence is altered; usually present in at
least 1% of the population. Also called SNP.
[Source – NCI Dictionary of Genetics Terms]
Singleton: typically used to refer to the sequencing of one individual rather than a family unit.
[Source – CSER Consortium Practitioner Education Working Group]
Toxicity: the dosage at which a drug causes adverse eects within the body.
[Source – CSER Consortium Practitioner Education Working Group]
Trio: typically refers to the sequencing and analysis of an aected individual as well has his or her
mother and father.
[Source – CSER Consortium Practitioner Education Working Group]
Variant calling: the process of comparing a DNA sequence of interest to a reference.
[Source – CSER Consortium Practitioner Education Working Group]
Variant of uncertain signicance: A variant of uncertain signicance is a change in the DNA
sequence whose association with disease is unknown. Also called a variant of unknown signicance,
unclassied variant, or sometimes simply a VUS.
[Source – CSER Consortium Practitioner Education Working Group]
Glossary

34
X-linked recessive: a mode of inheritance in which a mutation (variant) in a gene on the X
chromosome causes the phenotype (symptoms) to be expressed in males who are hemizygous for
the gene mutation (i.e., they have only one X chromosome) and in females who are homozygous for
the gene mutation (i.e., they have a copy of the gene mutation on each of their two X chromosomes).
Carrier females who have only one copy of the mutation do not usually express the phenotype,
although dierences in X-chromosome inactivation can lead to varying degrees of clinical expression
in carrier females.
[Source – Illustrated Glossary]
Glossary

