
The EMBO Journal Vol.16 No.24 pp.7444–7456, 1997
Cisplatin- and UV-damaged DNA lure the basal
transcription factor TFIID/TBP
Paul Vichi
1
, Fre
´
de
´
ric Coin,
Jean-Paul Renaud, Wim Vermeulen
2
,
J.H.J.Hoeijmakers
2
, Dino Moras
and Jean-Marc Egly
3
Institut de Ge
´
ne
´
tique et de Biologie Mole
´
culaire et Cellulaire, BP 163,
F-67404, Illkirch Cedex, Universite
´
Louis Pasteur, Strasbourg, France
and
2
Department of Cell Biology and Genetics, Medical Genetics
Center, Erasmus University Rotterdam, P.O. Box 1738,
3000 DR Rotterdam, The Netherlands
1
Present address: University of Vermont, Department of Molecular
Physiology and Biophysics, Burlington, VT 05405, USA
3
Corresponding author
P.Vichi and F.Coin contributed equally to this work
A connection between transcription and DNA repair
was demonstrated previously through the characteriz-
ation of TFIIH. Using filter binding as well as in vitro
transcription challenge competition assays, we now
show that the promoter recognition factor TATA box-
binding protein (TBP)/TFIID binds selectively to and
is sequestered by cisplatin- or UV-damaged DNA,
either alone or in the context of a larger protein
complex including TFIIH. Computer-assisted 3D struc-
tural analysis reveals a remarkable similarity between
the structure of the TATA box as found in its TBP
complex and that of either platinated or UV-damaged
oligonucleotides. Thus, cisplatin-treated or UV-irradi-
ated DNA could be used as a competing binding site
which may lure TBP/TFIID away from its normal
promoter sequence, partially explaining the phenome-
non of DNA damage-induced inhibition of RNA syn-
thesis. Consistent with an involvement of damaged
DNA-specific binding of TBP in inhibiting transcrip-
tion, we find that microinjection of additional TBP in
living human fibroblasts alleviates the reduction in
RNA synthesis after UV irradiation. Future anticancer
drugs could be designed with the consideration of
lesion recognition by TBP and their ability to reduce
transcription.
Keywords: cisplatin/DNA repair/TBP/TFIID/UV
irradiation
Introduction
Nucleotide excision repair (NER) is essential for the
genomic repair of UV-induced pyrimidine dimers or bulky,
helix-distorting chemical adducts caused by numerous
compounds such as acetylaminofluorene (AAF) or the
anticancer drug, cisplatin (Zamble and Lippard, 1995).
Early investigations aimed at elucidating how cells respond
to damage induced by such diverse agents demonstrated
a transcriptionally linked subpathway of NER in which
7444
© Oxford University Press
lesions in transcribed genes were repaired preferentially
(Bohr et al., 1985). A further bias was shown by the
increased removal of damage in the coding versus the
non-coding strand and was suggested to result from
participation of the arrested RNA polymerase II (RNA
pol II) elongation complex which might serve to recruit
repair machinery (Mellon et al., 1987; Leadon and
Lawrence, 1991; Sweder and Hanawalt, 1992).
More recently, it was established that the basal transcrip-
tion factor TFIIH, critical for transcription, was also an
intricate component of NER (for reviews, see Hoeijmakers
et al., 1996; Svejstrup et al., 1996). This dual function
suggests that TFIIH, as part of the transcription initiation
complex, is well positioned to assist in the rapid removal
of lesions in transcribed genes. What remains unclear is
how the TFIIH complex, an essential transcription factor
as well as part of the core NER machinery required for
both global and transcription-coupled repair, is shared
between these two distinct processes. Difficulties in under-
standing the regulation/function of TFIIH in repair versus
transcription reflect the involvement of numerous proteins
and their relative interactions in each process. This is
complicated further by differences in in vitro conditions
required to support each process. As a result, studies
concerned with the role of TFIIH during in vitro NER or
transcription are usually performed in the absence of one
process. However, the notion that tight connections exist
between NER and transcription, and the dual involvement
of components in both processes, prompted us to investi-
gate whether damaged DNA produces a high affinity site
which is able to sequester factors supporting NER and/or
transcription (Iyer et al., 1996). We test this hypothesis
by developing a challenge in vitro transcription assay
using an undamaged transcription unit as a template in
the presence of either UV-irradiated or cisplatin-damaged
DNA as a competitor. This assay still examines a selective
function of TFIIH (transcription) but, through pre-incub-
ation of damaged DNA with whole cell extracts or purified
factors, allows processes of NER to be initiatied and allows
the observation of the relative influence of interactions
between such factors on the ability of these extracts to
support transcription. Our results indicate that the presence
of damaged DNA leads to an inhibition of transcription
from an independent and transcriptionally viable template.
The most likely interpretation of this trans-effect on
transcription is the sequestration of TFIIH in repair events,
rendering it unavailable for transcription initiation. How-
ever, administration of extra TFIIH had only a relatively
minor effect on recovery of RNA synthesis. Unexpectedly,
addition of the recombinant TATA box-binding protein
TBP or the whole TFIID also appeared to largely abolish
the inhibition of transcription. TBP was found sub-
sequently to bind strongly to different types of damaged
DNA. Computer-assisted 3D structural analysis reveals a
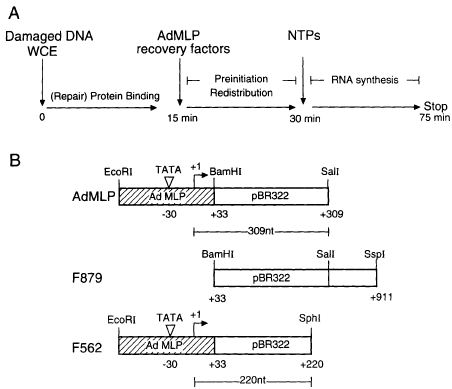
Damaged DNA lures transcription factor TBP/TFIID
Fig. 1. (A) Design of transcription competition experiment.
(B) Description of DNA fragments used for the in vitro transcription
competition assay. AdMLP and F562 transcription templates are
generated by EcoRI–SalI and EcoRI–SphI restriction digestion of
plasmid pUC309 and give rise to 309 and 220 nt RNA transcripts
respectively. Both contain the AdMLP promoter (hatched bars).
Fragment F879 was created by restriction digestion of pUC309 with
BamHI and SspI. This fragment does not contain any promoter
sequence.
remarkable similarity between the structure of the TATA
box as found in its TBP complex and that of platinated
oligonucleotides. Consistent with an involvement of
damaged DNA-specific binding of TBP resulting in the
inhibition of transcription, we also find that microinjection
of additional TBP into living human fibroblasts alleviates
the reduction in RNA synthesis after UV irradiation.
The sequestration of this crucial transcription factor by
DNA lesions could partially explain the overall reduction
in RNA synthesis observed in vivo after genotoxic treat-
ment, and thus represents part of the cellular response to
DNA damage.
Results
Addition of damaged DNA inhibits in vitro
transcription
The presence of DNA lesions recognized by TFIIH, alone
or in concert with other NER proteins, may reduce the
availability of TFIIH or other transcription factors, thereby
inhibiting the formation of a functional transcription
initiation complex. To investigate this possibility, we
developed a crude in vitro transcription competition assay
(Figure 1A).
HeLa whole cell extract (WCE) was pre-incubated with
various amounts of cisplatin- or UV-damaged DNA under
conditions which only allow the formation of the pre-
incision complex, one of the first steps of NER. The
following incision/excision and resynthesis steps are
inhibited by the low ATP concentration and the absence
of dNTP respectively (Calsou and Salles, 1994; Moggs
et al., 1996). After the first 15 min, an AdMLP reporter
template was introduced and the reactions were continued
for an additional 15 min to allow the formation of pre-
initiation transcription complexes and for any redistribu-
tion of factors including TFIIH between damaged DNA
7445
and transcription template. RNA synthesis was then initi-
ated by addition of NTPs and quantified by the production
of a 309 nt transcript. Pre-incubation of WCE with a
UV-irradiated 879 bp fragment (F879 UV1) (containing
~3–4 lesions per DNA molecule, Jones and Wood, 1993)
lacking promoter sequences (Figure 1B) inhibited tran-
scription from the AdMLP template (Figure 2A, compare
lanes 2–5 with lanes 6–9). Similarly, pre-incubation of the
3 kb pSK plasmid containing ~30 cisplatin-induced DNA
crosslinks (Hansson and Wood, 1989) also inhibited tran-
scription compared wirh undamaged DNA (Figure 2B,
compare lanes 2–8 with lanes 9–15).
Quantification of the synthesis of RNA transcript
(309 nt) from several experiments repeatedly shows that
both UV- and cisplatin-damaged DNA inhibit transcription
from an AdMLP template 3- to 4-fold more than un-
damaged DNA (see Figure 2A and B, lower panels). Note
that 50% transcription inhibition is observed with a 2-fold
excess of cisplatinated sites compared with TATA promoter
sites, in the conditions described in Figure 2B.
To ensure that inhibition of the 309 nt transcript reflected
a titration of factors on damaged DNA necessary to
support transcription, we included a fixed concentration
of a competitor DNA in the reaction but varied the
ratio of damaged to undamaged fragment. Under these
conditions, inhibition of transcription was observed as a
function of the increase in damaged fragment present
(Figure 2A, lanes 10 and 11). To demonstrate further that
inhibition resulted from the loss of critical transcription
factors on damaged DNA rather than non-specific protein–
DNA or DNA–DNA interactions, we performed transcrip-
tion competition experiments using two transcribable tem-
plates: the AdMLP reporter template (product 5 309 nt)
and a second AdMLP-containing template, F562, which
produces a 220 nt transcript (Davison et al., 1983). We
reasoned that if a decrease in the production of the 309 nt
transcript resulted solely from DNA–DNA or non-specific
protein interactions, then transcription from a transcribable
competitor may also be inhibited when presented in
an undamaged form. However, we observed that pre-
incubation of increasing amounts of UV-irradiated F562
resulted in a more effective inhibition of transcription from
the AdMLP template than pre-incubation with undamaged
F562 DNA (Figure 2C, compare the 309 nt transcript
from lanes 8–13 with lanes 2–7). The presence of un-
damaged F562 decreased transcription somewhat from the
AdMLP reporter template (309 nt), as might be expected
since transcription factors would now be shared between
two active promoters; however, the F562 template was
still capable of being transcribed (220 nt), suggesting that
the presence of two different DNAs in our reaction was
not leading to interactions which rendered the templates
unavailable for transcription. Only in the presence of UV-
irradiated F562 did we also observe the absence of the
220 nt transcript (Figure 2C, lanes 8–13). The lack of the
220 nt product demonstrates that transcription factor(s)
which are associated with damaged DNA (F562 UV1)
are unable to support transcription from that template but,
more importantly, are also unavailable to participate in
transcription from an independent, undamaged template
(AdMLP). Together with data presented in Figure 2A,
these experiments demonstrate that the reaction conditions
support transcription from two templates and that DNA
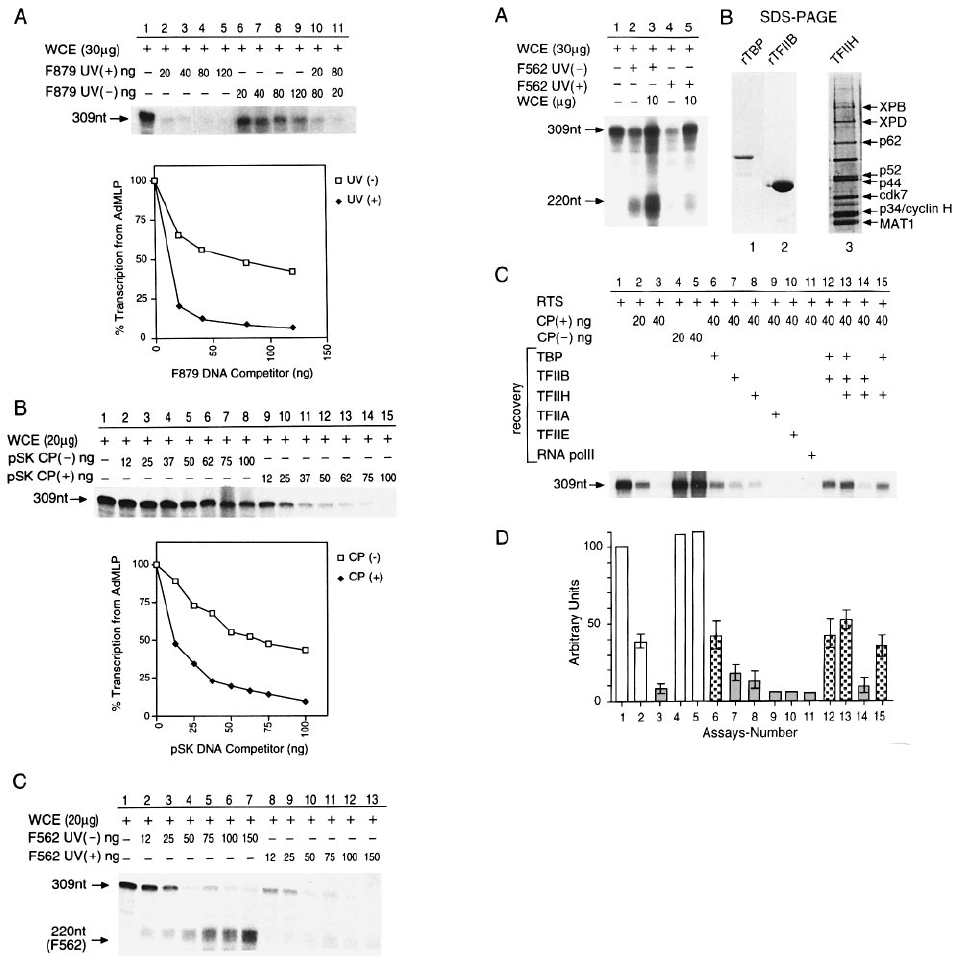
P.Vichi et al.
Fig. 2. Inhibition of in vitro transcription from the AdMLP by the
presence of damaged DNA. (A) Transcription of AdMLP (50 ng) is
performed with 30 µg of WCE in the presence of increasing amounts
of UV-irradiated (3 kJ/m
2
) (lanes 2–5) or non-irradiated (lanes 6–9)
F879 DNA fragment. Upper panel: autoradiogram; lower panel:
densitometric quantification of autoradiogram (309 nt band), presented
as the percentage of transcription from the AdMLP as a function of
the amount of F879 competitor DNA. Transcription in the absence of
competitor equals 100%. (B) Transcription of AdMLP (50 ng) is
performed with 20 µg of WCE in the presence of increasing amounts
of plasmid pSK CP(–) (lanes 2–8) or CP(1) (lanes 9–15). Top panel:
autoradiogram showing transcription from the AdMLP (309 nt);
bottom panel: graph representing quantification by PhosphoImage
analysis of the autoradiogram. (C) Inhibition of transcription by
UV-irradiated F562 DNA. Transcription of AdMLP is performed with
20 µg of WCE in the presence of increasing amounts of non-irradiated
(lanes 2–7) or UV-irradiated (1.5 kJ/m
2
) (lanes 8–13) F562 DNA
fragment. The size of each transcript is indicated.
containing UV- or cisplatin-induced lesions inhibits tran-
scription because it interacts with components absolutely
required for transcription from a TATA promoter.
7446
Fig. 3. Transcription factors are associated with damaged DNA.
(A) UV-irradiated (1.5 kJ/m
2
) (25 ng) or non-irradiated (25 ng) F562
DNA fragment was incubated as described in Figure 2C with 30µgof
WCE. Recovery was obtained with the addition of 10 µgofWCE
(lanes 3 and 5) to reactions inhibited by non-irradiated and
UV-irradiated F562 DNA respectively. (B) Purified recombinant TBP
and TFIIB as well as purified TFIIH from HeLa cells (hydroxyapatite
fraction, Humbert et al., 1994) were analysed by SDS–PAGE.
(C) Transcription of AdMLP (50 ng) was carried out in a highly
purified reconstituted in vitro transcription system (RTS). Cisplatin-
damaged pSK DNA is pre-incubated with all components of the RTS
before addition of AdMLP and either of the various transcription
factors indicated at the top of the figure. The added amount of factors
being tested for their ability to restore transcription was the same as
that used to assemble the reconstituted assay, effectively doubling the
concentration during the recovery. (D) The scanned transcription
activities of (C) are reported together with values of two other
experiments. In the shaded and chequered columns, 40 ng of damaged
DNA is added; the chequered columns contain TBP. The means 6 SE
are indicated.
TBP is sequestered by DNA damage
In order to determine the specific transcription factors
contributing to the loss of activity in the presence of
damaged DNA, we carried out transcription competition
experiments in which WCE or purified transcription factors
were added to the transcription reaction together with the
AdMLP fragment. Addition of WCE increases transcrip-
tion from either AdMLP (309 nt) or F562 (220 nt) (Figure
3A, compare lanes 2 and 3 with lanes 4 and 5). When
added to a reaction inhibited by UV-damaged DNA, WCE
significantly restored the level of transcription (compare

Damaged DNA lures transcription factor TBP/TFIID
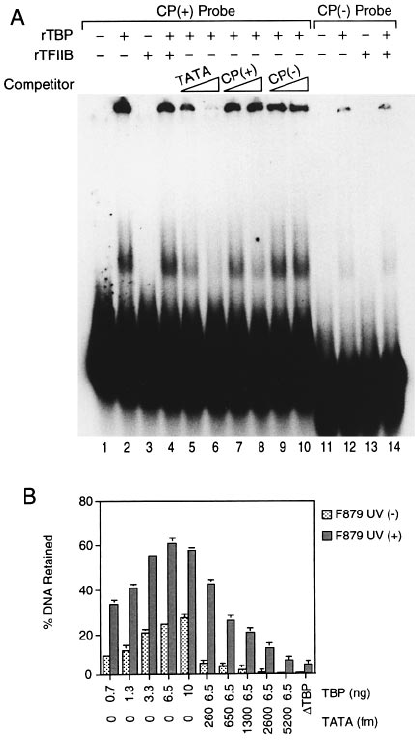
Fig. 4. Preferential binding of TBP to damaged DNA. (A)
32
P-Labelled F879 DNA was UV irradiated at different doses as indicated in the right part
of the panel, incubated with various amounts of TBP and tested for its retention on nitrocellulose filters. Graphs represent the percentage of DNA
retained on the filters as a function of the amount of TBP for four different doses of UV and for an undamaged F879 DNA fragment. Quantification
was performed using a PhosphoImage analyser; 100% represents the total counts obtained when 1 µl of each DNA probe (input) was spotted onto
Whatman paper and simultaneously exposed with the nitrocellulose filter. (B) Inhibition of AdMLP-dependent transcription in a highly purified
reconstituted in vitro transcription system is dependent on the UV dose. The reconstituted transcription assay was carried out in the presence of
increasing amounts of non-irradiated (lanes 2–5), 500 J/m
2
(lanes 6–9) and 1000 J/m
2
(lanes 10–13) UV-irradiated F879 DNA. (C) The transcription
reaction was carried out in an RTS with F879 UV (1) (1000 J/m
2
) or (–) DNA probe as competitor. Recovery of transcription after inhibition with
competitor DNA was obtained by adding TBP. (D) The filter binding assay was performed as described in (A), except that a PvuI restriction
fragment (1084 bp) of pSK cisplatin-treated CP(1) or untreated CP(–) was used as the DNA probe. (E) The transcription reaction was carried out in
an RTS with an increasing amount of cold CP(–) or CP(1) pSK PvuI DNA probe (see D) as competitor. Recovery of transcription after inhibition
with CP(1) DNA was achieved by adding various amounts of TBP. (F) The transcription reaction was carried out as in (E) except that purified
TFIID was used in place of TBP. Recovery of transcription was obtained by adding various amounts of TFIID roughly corresponding to 6–15 ng of
TBP according to Western blot analysis.
lanes 1, 4 and 5). The weak 220 nt product from the F562
UV1 template (lane 5) is either non-specific or results
from residual undamaged template. Together, these results
demonstrate that our assay is responsive to new factors
and that we are below saturating conditions with respect
to total protein (compare lanes 2 and 3 with lanes 4 and
5). Furthermore, these results also demonstrate that UV
lesions in the damaged F562 template remain a significant
block to transcription even in the presence of additional
proteins (compare lanes 3 and 5).
Besides TFIIH, WCE contains many other factors which
may contribute to the observed restoration of transcription.
In addition, controlling the relative amounts of individual
factors within a WCE is virtually impossible. We therefore
used a reconstituted transcription system (RTS) in order
to determine effectively the role of each transcription
factor. Thus in vitro transcription competition experiments
were performed with an RTS containing highly purified
TBP, TFIIB, TFIIE, TFIIF, TFIIH, TFIIA and RNA pol
II (Humbert et al., 1994; see also Figure 3B). The RTS
was pre-incubated with cisplatin-damaged DNA, and the
ability of each purified transcription factor to restore
activity was determined. Addition of TBP, and to a lesser
extent addition of TFIIB and TFIIH, restored transcription
(compare Figure 3C and D, lane 3, with lanes 6–8),
7447
whereas addition of TFIIA, TFIIE or RNA pol II had no
effect (lanes 9–11).
However, the addition of TFIIH and TFIIB, either alone
or in combination with TBP, did not lead to a significantly
greater degree of restoration than that provided by TBP
itself, suggesting that this was the limiting primary factor
after incubation of the RTS with damaged DNA. In fact
addition of increasing amounts of either TBP or TFIID,
in an RTS already inhibited by cisplatin-treated DNA
(Figure 4E and F), was able to restore activity to the
initial level of transcription without inducing similar
increases in the reaction performed in the absence of
competitors (Figure 4E, compare lane 1 with lanes 8 and
9). Together these results suggest that TBP interacts
directly with the DNA lesion, whereas TFIIH and TFIIB
may either be associated with DNA lesions through the
TBP, or drive transcription through stabilization of the
TBP–promoter complex (see Discussion and Ge
´
rard et al.,
1991). Furthermore, TBP and TFIIB as well as purified
TFIIH were shown to be free of repair proteins (Figure
3B; see also Aboussekra et al., 1995), indicating that
binding to damaged DNA as well as recovery of transcrip-
tion is not the result of a contaminating repair factor.
Although TFIIH has been reported to be recruited to the
DNA lesion site in conjunction with other factors (Park
P.Vichi et al.
et al., 1995), the added presence in our reconstituted
transcription system of recombinant xeroderma pig-
mentosum group A (XPA) protein did not significantly
affect transcription (data not shown).
TBP interacts directly with damaged DNA
The ability of TBP, TFIIB and TFIIH to restore transcrip-
tion inhibited by the presence of damaged DNA suggested
that these factors may interact with DNA lesions. We
investigate this possibility using a standard nitrocellulose
filter binding assay. As illustrated in Figure 4A, treatment
of F879 DNA, which does not contain a TATA element
(Figure 1B), with increasing doses of UV irradiation
(Figure 4B; 100–1500 J/m
2
) resulted in a corresponding
increase in the amount of DNA retained by a fixed
concentration of TBP. The functional significance of
this interaction is demonstrated further in the following
experiment. The RTS, containing all of the basal transcrip-
tion factors, was pre-incubated with F879 DNA fragment
damaged by irradiation with either 500 or 1000 J/m
2
, and
assayed for its ability to support transcription from the
AdMLP reporter template. In agreement with observations
from the crude transcription assay and nitrocellulose
filter binding assays, the presence of damaged DNA
preferentially inhibited the production of the 309 nt
transcript (Figure 4B, compare lanes 2–5 with 6–9 and
10–13). The inhibition can be reversed by back addition
of TBP (Figure 4C).
TBP also recognizes cisplatin-damaged DNA, as judged
by the nitrocellulose filter binding assay (Figure 4D). Pre-
incubation of the RTS containing recombinant TBP with
cisplatin-damaged DNA resulted in a specific inhibition
of transcription from the AdMLP template (Figure 4E,
compare lanes 2 and 3 with lanes 4 and 5). Transcription
can be restored completely after readdition of TBP (lanes
6 and 7). It is worthwhile noting that our assay is saturated
with respect to all individual components for a given
concentration of AdMLP template. For example, we
noticed that addition of TBP did not result in a general
increase in the level of transcription (Figure 4E, compare
lane 1 with lanes 8 and 9). When the same experiment
was performed with an RTS containing TFIID (the native
transcription factor that contains TBP) instead of TBP,
cisplatinated DNA also inhibited the transcription reaction
(Figure 4F, lanes 1–5); here too, transcription is restored
upon addition of an excess of TFIID (lanes 6–7), thus
demonstrating that in the context of TFIID, TBP can still
bind to damaged DNA.
TFIIB, which was also able to restore transcription, was
unable to retain a damaged or undamaged fragment
(data not shown), suggesting that its contribution to the
inhibition/restoration of transcription is indirect, most
probably through stabilization of TBP on the promoter.
Attempts to use filter binding assays to demonstrate a
specific association of highly purified TFIIH alone with
damaged DNA were also unsuccessful. Highly purified
TFIIH retained, although weakly, similar levels of both
damaged and undamaged DNA, suggesting that binding
to damaged DNA probably occurs through interactions
with other repair factors such as XPA (Park et al.,
1995) or the transcription factor TBP, as suggested by
transcription recovery experiments.
The ability of purified TBP to bind to damaged DNA
7448
Fig. 5. The TATA box-containing fragment competes with damaged
DNA. (A) Gel shift analysis of rTBP binding to cisplatin-damaged
DNA. Binding was performed with the indicated cisplatin-damaged
CP(1) or undamaged CP(–) 36mer DNA probe (0.5 ng; 10 000
c.p.m.), rTBP (20 ng) and/or rTFIIB (20 ng). Then 10 and 50 ng of a
64mer AdMLP fragment containing the TATA box (TATA) or 10 and
50 ng of PvuI pSK CP(1) and CP(–) DNA fragment were added as
competitors as indicated. Note that the radioactive material in the
wells is due to recombinant TBP–DNA probe aggregates.
(B) Inhibition of TBP binding to UV-irradiated (1.5 kJ/m
2
) F879 DNA
was performed by addition of increasing amounts of the TATA DNA
fragment (64mer) also used in the EMSA. When heat inactivated
(47°C), TBP interacts weakly with DNA.
was also observed in standard electrophoresis mobility
shift assays (EMSAs). A
32
P-labelled DNA probe, either
undamaged or containing a single 1,3-GpTpG cisplatin
crosslink, was incubated with rTBP and non-specific DNA
(see Materials and methods), and complex formation was
detected by a change in the migration pattern of DNA on
acrylamide gels. EMSA revealed the formation of a TBP–
DNA nucleoprotein complex that was specific for damaged
DNA (Figure 5A, lanes 2 and 12). Moreover, the formation
of a TBP–damaged DNA complex was reduced specifically
with increasing concentrations of an unlabelled cisplatin-
damaged PvuI pSK DNA fragment as compared with a
non-damaged fragment (compare lanes 7 and 8 with
lanes 9 and 10). The addition of an unlabelled fragment
containing a TATA box (TATA) alsoresulted in competition
with TBP binding to the cisplatin-damaged DNA probe
(compare lane 2 with lanes 5 and 6), supporting conclu-

Damaged DNA lures transcription factor TBP/TFIID
Table I. Effect of TBPr injection on transcription in living human fibroblasts
Exp. Microinjected UV irradiation Incubation RNA synthesis
c
DNA repair
d
sample
a
(J/m
2
) time (h)
b
(grains/nucleus) (grains/nucleus)
1 400 ng/µl rTBP 16 1 20.0 6 1.0
Non-injected 16 1 21.0 6 1.0
2 400 ng/µl rTBP 0 1 12.0 6 1.02
40 ng/µl rTBP 0 1 14.0 6 1.0
4 ng/µl rTBP 0 1 21.0 6 1.0
Non-injected 0 1 22.0 6 1.0
3 16 ng/µl rTBP 0 1 74.0 6 2.0
Non-injected 0 1 73.0 6 2.0
16 ng/µl rTBP 16 1 54.0 6 3.0
Non-injected 16 1 34.0 6 1.0
4 16 ng/µl rTBP 16 3 43.5 6 3.5
50 ng/µl TFIIB 16 3 27.8 6 1.0
Non-injected 16 3 27.0 6 2.5
5* 16 ng/µl rTBP 16 3 42.0 6 3.0
Non-injected 16 3 25.0 6 1.0
6 16 ng/µl rTBP 30 3 34.0 6 1.0
Non-injected 30 3 19.0 6 1.0
a
Injections were performed in control fibroblasts (C5RO), except for Exp 5 where fibroblasts of a CS-B patient (CS1AN) are injected.
b
Time of incubation after UV irradiation and before pulse labelling with radioactive nucleotides.
c
RNA synthesis (with and without UV challenge), autoradiographically measured by [
3
H]uridine incorporation (Vermeulen et al., 1994) expressed as
grains/nucleus 6 SEM (at least 50 nuclei for each sample are counted).
d
UV-induced DNA repair synthesis (UDS), measured autoradiographically as the [
3
H]thymidine incorporation, expressed as grains/nucleus 6 SEM
(.50 nuclei/sample).
*CS-B cells also presented in Figure 6B.
sions that the shifted probe corresponded to a TBP–DNA
complex. TFIIB alone was unable to shift the damaged
DNA probe, and no supershift was observed when TFIIB
was added to the TBP–damaged DNA complex (see lanes
3 and 4 respectively). These conclusions were supported
by additional filter binding assays assessing the relative
affinity of TBP for a DNA lesion in the presence of the
normal TATA sequence. UV-irradiated F879 DNA (Figure
1B) was incubated with TBP in the presence of the TATA
element of the AdMLP (TATA). The results illustrated in
Figure 5B confirm a preferential association of TBP with
damaged DNA (column sets for TBP concentration 0.7–
10 ng) and indicate that significantly greater amounts of
the TATA fragment were required to compete the binding
of TBP on UV-damaged DNA to similar levels to that
observed with undamaged DNA (column sets for TATA
competitor, concentration 260–5200 fmol). A rough
estimation indicates that ~650 fmol of TATA box-con-
taining fragment (~200-fold) is necessary to achieve a
50% competition inhibition of the association of highly
purified TBP with the 3–5 fmol of damaged sites induced
by UV irradiation (according to Jones and Wood, 1993)
(see also Figure 3C). As a control, inactivated TBP,
previously incubated for 15 min at 47°C (Nakajima
et al., 1988), shows no interaction with UV-damaged or
undamaged DNA. Together, these results indicate for the
first time that two types of DNA lesions, induced by either
UV irradiation or cisplatin treatment, serve as binding
targets for TBP.
TBP microinjection protects cells from UV-induced
inhibition of RNA synthesis
UV-induced DNA damage in the genome causes a transient
inhibition of overall transcription. One possibility is that
this suppression is at least in part due to sequestration of
TBP by DNA lesions. Therefore, we reasoned that micro-
7449
injection of extra TBP might partly relieve this UV-induced
inhibitory effect. To test this possibility, rTBP was micro-
injectedintonormalprimary fibroblasts.TheeffectonNER,
transcription and UV-induced inhibition of transcription
was assessed by incubating the cells after rTBP injection,
in the presence of
3
H-labelled thymine (for repair synthesis)
or
3
H-labelled uridine (to measure transcription). NER and
transcription were quantified by counting the number of
autoradiographic grains above the nuclei of injected cells
and compared with the non-injected cells on the same slide.
Initial experiments (Table I, Exp 1 and 2; using rTBP at a
concentration of 400 ng/µl)indicated that the injected rTBP
by itself did not affect DNA repair (Exp 1) but caused a
strong inhibition of transcription (Exp 2). Apparently, the
excess TBP (we calculate that we injected ~40–60% of the
total cellular TBP content) squelches factors interfering
with normal transcription, a fact that was also observed
after transient overexpression of TBP (S.Buratowsky and
P.Chambon, personal communications). To avoid a domin-
ant-negative effect on regular transcription, we titrated TBP
to a concentration at which no inhibition was observed (4
and 16 ng/µl, see Exp 2 and Exp 3 respectively). As shown
in Table I (Exp 3, 4 and 6), injection of 16 ng/µl rTBP
induced a clear protective effect against the inhibition of
transcription caused by different doses of UV irradiation
(see also Figure 6A). The partial relief of transcription was
observed 1 and 3 h after UV exposure and was independent
of transcription-coupled repair, as injection in Cockayne
syndrome (CS)-B fibroblasts (which exhibit a selective
defect in transcription-coupled repair) also stimulated UV-
suppressed RNA synthesis (Exp 5 and Figure 6B). Micro-
injection of TFIIB (50 ng/µl, Exp 4) and cdk7–cyclinH–
MAT1 complex (50 ng/µl) failed to reverse the UV-induced
transcription inhibition, indicating that the observed protec-
tion was TBP specific. These findings showthat at least part
of the suppression of transcription by UV can be overcome
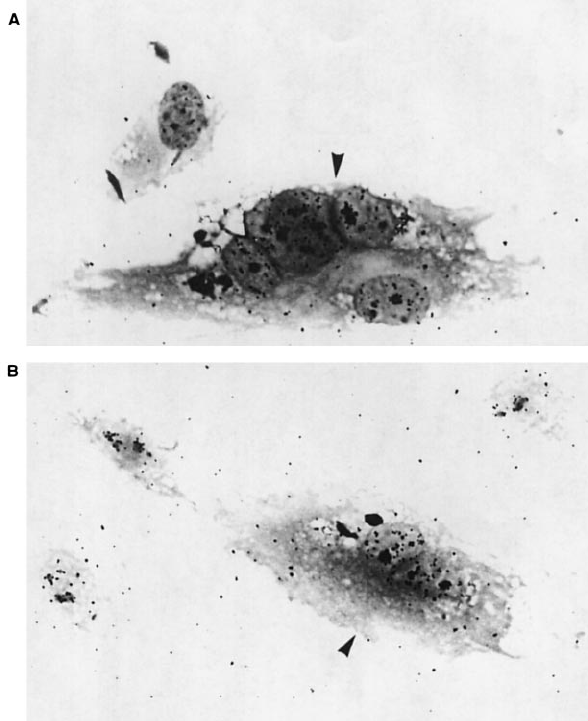
P.Vichi et al.
Fig. 6. Effect of TBP on transcription in vivo. Micrograph of C5RO normal (A) and CS-B patient (B) fibroblasts injected (indicated by an arrow)
1 h before UV irradiation (16 J/m
2
) showing an increase in the overall RNA synthesis. In each panel, one has to compare the autoradiographic
grains above the nuclei of injected cells (indicated by an arrow) with the surrounding nuclei.
by exogeneous TBP. This is consistent with the idea that
after UV irradiation, sequestration of endogeneous TBP
to DNA lesions takes place, contributing to the damage-
induced inhibition of transcription.
Similarities between the TATA box and
cisplatin-damaged DNA
We investigated the structural implications of these observ-
ations by comparing the structure of a platinated DNA
with its TATA counterpart from the human TBP–TATA
complex. The only available crystal structure of a 1,2-
cisplatin adduct on an oligonucleotide is that of a double-
stranded DNA dodecamer containing a central G∧G site
(cis-[Pt(NH
3
)
2
-{d(GpG)-N7(G
6
), N7(G
7
)}] intrastrand
crosslink) (Takahara et al., 1995), hereafter referred to as
GGPG. Superposition of GGPG onto the TATA box
DNA (TATA) revealed a strikingly similar overall shape,
especially in the central region, although the detailed
conformations of the base pairs differ in the two structures
(Figures 7A and 8A and B). The following analysis is
based on the optimal superposition of the original structural
data without any energy minimization. An r.m.s.d. of 2.1
Å was found between the backbone atoms of TATA and
GGPG (compared with an r.m.s.d. of 8.8 Å between the
backbone atoms of TATA and BDNA, the canonical
B-DNA dodecamer, and an r.m.s.d. of 5.7 Å between the
7450
backbone atoms of GGPG and BDNA, compare Figure
7B and C). To calculate a realistic r.m.s.d. value, we have
taken into account the different orientation of the base
pairs in the two structures which leads to a shift of one
nucleotide on one strand (Figure 7A). This explains how
the two overall structures can fit so well in spite of
important differences in base pair conformations. Both
TATA and GGPG are bent towards the major groove and
partially unwound without disrupting the Watson–Crick
hydrogen bonding pattern. In the TBP–TATA complex,
the saddle-shaped TBP core wraps around the DNA in
the minor groove (Chasman et al., 1993; J.L.Kim et al.,
1993; Y.Kim et al., 1993; Juo et al., 1996). The pronounced
bend is induced by the insertion of phenylalanine side
chains into the first and last steps of the TATA element,
and is favoured by the intrinsic bendability of the TA
steps. In the GGPG structure, a similar bend is produced
by the coordination of the platinum ion to the N7 nitrogen
atoms of two adjacent guanines on the same strand, which
forces the destacking of the complementary bases. As a
result, the cisplatin adduct mimicks the distorted conform-
ation of TATA in its complex with TBP. In both molecules,
the structure exhibits an abrupt B- to A-form transition
near the bent portion, with a pronounced opening and
flattening of the minor groove, as illustrated in the two by
two superpositions of TATA, BDNA and GGPG (Figure 7).
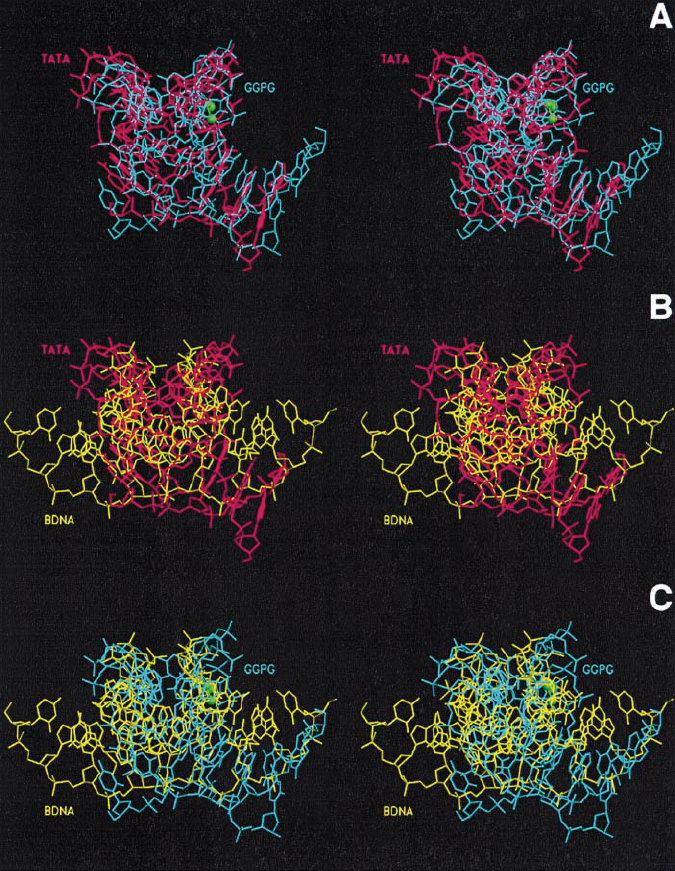
Damaged DNA lures transcription factor TBP/TFIID
Fig. 7. Stereoviews of the two by two superpositions of the crystal structures of the TATA box from the human TBP–TATA complex (TATA, PDB
code 1TGH; Juo et al., 1996), of the DNA dodecamer containing a central cis-[Pt(NH
3
)
2
-{d(GpG)-N7(G
6
), N7(G
7
)} intrastrand crosslink (GGPG,
PDB code 1GPG; Takahara et al., 1995), and of a canonical B-DNA dodecamer (BDNA, PDB code 1BNA; Drew et al., 1981). The superpositions
were optimized using the LSQ options of O (Jones et al., 1991) and displayed with SETOR (Evans, 1993). TATA is shown in red, GGPG in cyan,
with the platinum ion and the nitrogen atoms of its amine ligands in green, and BDNA in yellow. (A) GGPG–TATA; (B) BDNA–TATA;
(C) BDNA–GGPG.
Indeed, TBP appears to dock exceedingly well on
GGPG (Figure 8A and B), the complex showing very
few stereochemical clashes at the level of the inserted
phenylalanines. An optimal fit would require a small
adaptation of DNA with a minimal energy cost. The
buried surface in the TBP–GGPG complex calculated
from the docked structures is 2707 Å
2
compared with
3090 Å
2
in the TBP–TATA complex, the slightly lower
value arising from the more pronounced bend in GGPG,
as seen in Figure 8A. Note that the complex with GGPG
has not been energy-minimized. In both cases, the contacts
with TBP are essentially hydrophobic, with only a few
exceptions such as the hydrogen bond bridges formed by
Asn163 and Asn253 with the two central T
.
A base pairs
in the TBP–TATA complex. When BDNA is docked with
TBP, the interface drops down to 2241 Å
2
, and the
7451
minor groove forms a cavity which suppresses the contacts
between the protein side chains and the bases. Instead,
phosphate groups point towards the protein surface, making
the binding of TBP to DNA in a B conformation very
unfavourable (Figure 8C). The importance of the inter-
actions with the bases is supported by the observation that
two TBP mutants at the 253 position are defective in DNA
binding (Arndt et al., 1995; Lee and Struhl, 1995). As a
consequence, the helical twist is nearly zero at the central
step, the two base pairs being stacked directly on top of one
another with a 25° roll angle (Juo et al., 1996). This value
can be compared with the 26° roll angle observed between
the two central G*C base pairs in GGPG (Takahara et al.,
1995). Accordingtoourdockingexperiment,thetwoaspar-
agine residues are accommodated at the TBP–GGPG inter-
face, providing different hydrogen bonding patterns,
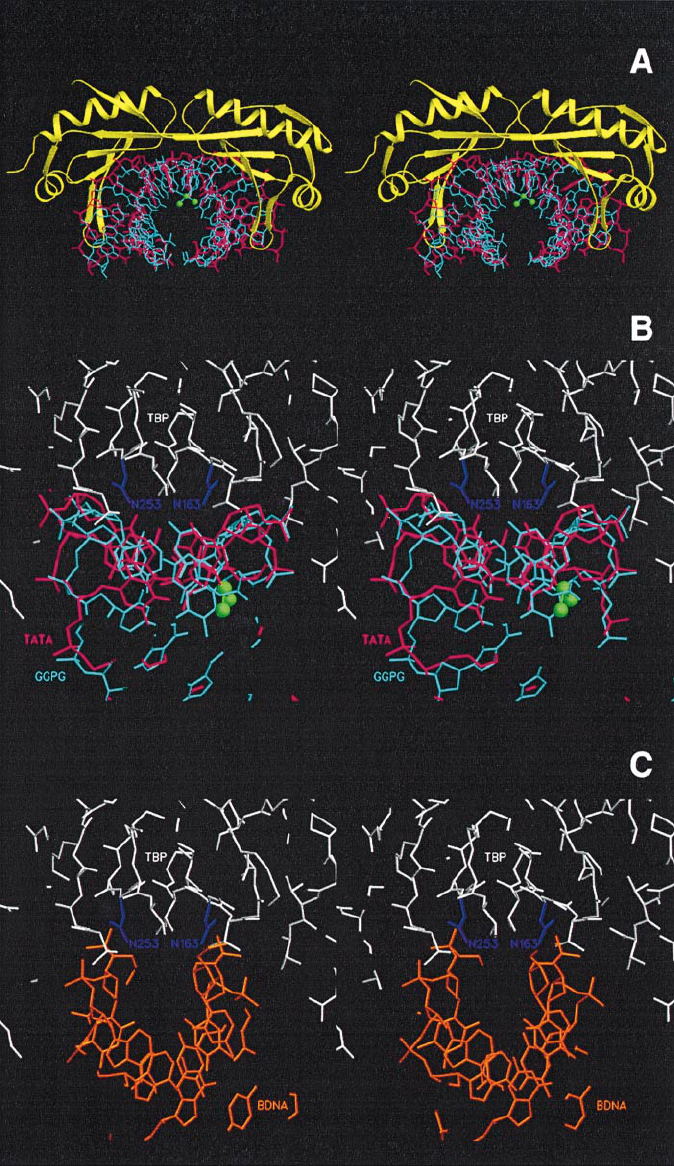
P.Vichi et al.
Fig. 8. Stereoviews of the TBP–DNA interfaces displayed with SETOR. (A) View along the long axis of TBP of the human TBP–TATA complex
showing GGPG superposed to TATA as in Figure 6A. TATA is shown in red, GGPG in cyan, with the platinum ion and the nitrogen atoms of its
ammine ligands shown in green, and TBP is displayed in yellow in a ribbon representation. (B) Close view in the same orientation as in Figure 6
showing TBP (in white) sitting on the minor groove. The Asn163 and Asn253 side chains are coloured in magenta. (C) View of BDNA docked with
TBP in the same orientation as in Figure 7B, showing the cavity at the interface and the unfavourable orientation of the central phosphate groups.
Asn163 and Asn253 contacting N3 of G
6
and O2 of C
19
,
respectively. Thus these interactions do not discriminate
between different sequences, but rather are involved in the
recognition of a particular type of DNA structure.
7452
Discussion
We present data which suggest that repair and transcription
may be linked at a more fundamental level than previously
Damaged DNA lures transcription factor TBP/TFIID
thought. Earlier work suggested a role for an arrested
RNA pol II complex at a DNA lesion (Donahue et al.,
1994), followed by participation of TFIIH in the recogni-
tion and incision/excision of the DNA damage, leading to
preferential repair (Moggs et al., 1996). Our work pre-
sented here suggests that TBP/TFIID, an essential com-
ponent which nucleates the formation of an active
transcription complex, recognizes and binds directly to
DNA lesions induced by UV irradiation or cisplatin
treatment.
TFIID/TBP directly binds damaged DNA
Using in vitro transcription challenge competition assays,
we show that TBP/TFIID, either in the context of a crude
cellular extract (WCE) and therefore in the presence of
all repair proteins, or in the presence of all the general
transcription factors and RNA pol II, associates with
damaged DNA. This association appears to be relatively
rapid and persistent, since changing either the time of pre-
incubation of WCE with damaged DNA or increasing the
pre-initiation time in the presence of both the competitor
and AdMLP DNA had little effect on the overall level of
inhibition induced by damaged DNA (data not shown).
The so-called TATA-binding protein, TBP, either free or
associated with the TBP-associated factors, TAFs (named
TFIID), directly interacts with damaged DNA as demon-
strated by nitrocellulose filter binding, gel shift and tran-
scription competition experiments. This was also observed
in reactions containing a TATA box, indicating that a
lesion can bind TBP efficiently even in the presence of
its specific (and natural) binding site. Moreover, the
strength of interaction of TBP with either UV-, cisplatin-
(the present study) or AAF- (unpublished results) damaged
DNA will be a function of the nature of the damage and
the surrounding sequences, which will each contribute to
the overall distortion in the DNA helix.
Transcription challenge competition assays show that
not only TBP but also TFIIB and TFIIH, although to a
lower extent, are required to restore AdMLP transcription
activity previously inhibited by the presence of a damaged
DNA fragment (Figure 3B). This is not particularly
surprising in light of the fact that TFIIH contains subunits
with zinc finger motifs (Humbert et al., 1994; data not
shown) and has been shown to interact with TBP (Ge
´
rard
et al., 1991). However, attempts to demonstrate a specific
association, using gel shift experiments, between TFIIH
or TFIIB alone or in combination with TBP and damaged
DNA were unsuccessful (data not shown). TFIIH recruit-
ment to the excision complex was shown to occur through
other factors present in the crude cellular extract (Nocentini
et al., 1997). It is possible that the association of TFIIB
and TFIIH with a DNA lesion or with a TBP-damaged
DNA complex could be rather weak compared with the
interactions which are required during the formation of
an active transcription complex. Partial restoration by
TFIIB and TFIIH may therefore reflect some interactions
with DNA lesions or the ability of these factors to disrupt
binding of TBP to damaged DNA. If TBP alone exhibits
a stronger affinity for damaged DNA, rather than for its
natural TATA target, addition of TFIIB or TFIIH may
stabilize existing binary TBP–TATA box complexes, dis-
placing the thermodynamic equilibrium away from the
damaged DNA complex towards the formation of a
7453
functional transcription initiation complex. The inability
of TFIIA, previously shown to stabilize TBP–TATA box
interactions (Buratowski et al., 1989), to recover transcrip-
tion inhibited by damaged DNA competitor may reflect
this being exclusively a promoter function. It also must
be borne in mind that the damaged cisplatin structure is
not completely identical to the TATA box structure and
thus may not be targeted equally by all the basal transcrip-
tion factors, e.g. TFIIA, TFIIB and TFIIH.
In vivo evidence for TBP/TFIID binding to damaged
DNA
To investigate a possible in vivo role of TBP/TFIID in
the cellular response to DNA damage, we performed
microinjection into living cells. The absence of an effect
on UV-induced DNA repair synthesis, under conditions
in which transcription was strongly inhibited, indicates
that both processes, although requiring the participation
of common factors such as TFIIH, are largely independent.
Furthermore, the lack of squelching by excess TBP on
NER suggests that TBP does not interact with essential
NER factor(s), and is therefore not implicated directly in
the NER process. However, microinjection of a well-
defined concentration of TBP was found to protect cells
from the reduction in transcription (overall RNA synthesis)
caused by UV exposure, whereas microinjection of either
TFIIB or the three components of the CAK complex
(Rossignol et al., 1997) had no significant effect. One
possible explanation for the relief provided by TBP is that
when exogeneously added, this protein binds directly to
lesions that would otherwise have trapped endogenous
transcription-competent TFIID or other SL1 or TFIIIB
complexes (two transcription factors which include TBP
and are associated with RNA pol I and RNA pol III
transcription respectively). The result of microinjection
then is to increase the pool of TBP-containing complexes
such as TFIID (Colgan and Manley, 1992) which would
be available for damage and/or promoter recognition. This
conclusion is supported by our filter binding studies. The
fact that TFIIB microinjection did not relieve transcription
inhibition may reflect the weak affinity of TFIIB for the
damage and/or its preference for the transcription reaction
in the context of an in vivo situation in which TFIIB plays
a crucial role in the activation process. Although the
microinjection experiments are not simple to interpret,
they fit well with our model in which TBP/TFIID binding
to lesions is at least partly responsible for the general
drop in transcription exerted by UV irradiation and other
DNA-damaging treatments.
TBP recognizes a typical 3D structure
The almost perfect match between the TBP core, as found
in its complex with the TATA box, and GGPG strongly
supports the present results of a specific binding of TBP
to cisplatin 1,2-adducts. In the former case, the TBP–
TATA interaction is an induced fit, while in the latter,
TBP seems to bind to a pre-formed, bent DNA in a lock-
and-key fashion. As noted by Juo et al. (1996), TBP
recognizes the intrinsic bendability of the TATA box more
than the base pair sequence per se. It is the opening of
the minor groove induced by the protein which allows a
snug fit of the concave surface of TBP against DNA. In
the 1,2-cisplatin G∧G adduct, the minor groove is already
P.Vichi et al.
exposed, inviting TBP. The limited number of polar
interactions with the bases, as discussed above, discards
any strong sequence specificity and favours a structural
recognition. As for the relationship with the UV-damaged
DNA, it is of interest to note that the crystal structure of
an oligonucleotide containing a cyclobutane-type thymine
dimer in complex with T4 endonuclease V also exhibits
a sharp kink around the thymine dimer portion, splitting
the duplex into two halves of B-DNA with a 60° inclination
between the two helical axes (Vassylyev et al., 1995), i.e.
two 30° bends. In this case too, the DNA shape is very
reminiscent of that of TATA bound to TBP, although the
thymine dimer undergoes different constraints from its
interaction with the endonuclease, including the flipping-
out of the adenine base complementary to the 59-thymine
of the dimer, which allows the excision process to take
place. The structural similarity of the deformation indicates
that this type of UV-damaged DNA could also bend
towards the major groove through interaction with TBP,
due to the instability of the TT step introduced by the
crosslinking of the two adjacent thymines.
It thus appears that various genotoxic and antitumour
agents could turn GC-rich sequences into potential sites for
TBP. This structural correlation supports the experimental
observation that several damaging factors have similar
effects, suggesting that in all cases the DNA bendability,
with a marked tendency to adopt locally an A-form
conformation with a flat, widened minor groove, is the
common property.
Implications of TFIID/TBP binding to damaged
DNA
The structural similarities between the TATA box and
DNA lesions implies an important basal role for TBP.
Indeed, TBP, either directly or within the context of one
of the multiprotein complexes SL1, TFIID and TFIIIB,
allows the initiation of transcription from the three classes
of promoters. The damage caused by agents such as UV
irradiation and cisplatin treatment results in an altered 3D
structure of the DNA similar to the one adopted by the
TATA sequence. These different lesions, forming a kind
of TATA-like 3D structure, may then be recognized by
TBP, with functional implications; they may serve as a
lure for TFIID/TBP, diverting it from its natural promoter
target, explaining the loss of transcription observed in
cells after DNA damage. In addition, binding of TBP to
damaged DNA could also serve to alter the equilibrium
of TFIIH associated with transcription or repair complexes.
Less TBP bound to promoter sequences would result in a
decrease in the number of pre-initiation complexes to
which TFIIH may be recruited and would lead to an
increase in the availability and association of TFIIH with
repair proteins. In this manner, TBP may stimulate the
repair function of TFIIH indirectly. However, such a
hypothesis does not exclude a possible role, if any, for TBP
in the first step of NER, simply through the recognition of
the lesion in conjunction with XPA and RPA, and the
recruitment of TFIIH. It remains to be determined how
TBP damage recognition leads to a decrease in overall or
selective transcription, resulting in apoptosis. Binding of
TBP could shield the lesion from repair proteins unless it
can be translocated efficiently. Furthermore, the persistent
presence of bound TBP may be responsible for the
7454
increased cytotoxicity of DNA-damaging agents in CS
cells (Mayne and Lehmann, 1982).
Although this kind of molecular decoy has been pro-
posed previously [hUBF, a transcription factor involved
in rRNA synthesis, was shown to be hijacked by cisplatin
adducts (Treiber et al., 1994)], this is the first time that a
functional consequence of this type of interaction
(hijacking) has been demonstrated. Depending on the
outcome, future anticancer drugs could be designed with
the consideration of lesion recognition by TBP, taking
into account the specific type of lesion, its affinity for
TBP and its tendency to compete transcription.
Materials and methods
Materials
HeLa WCEs, as well as all the components of the in vitro reconstituted
transcription system, were as described in Humbert et al. (1994).
Substrates used for in vitro transcription or filter binding analysis
were generated by restriction digestion of either pUC309 or pSK plasmid
DNA. pUC309 was created by ligation of an EcoRI–BamHI fragment,
corresponding to sequences –677 to 133 of the AdMLP (∆ –372/–34),
to the BamHI–SalI fragment from pBR322. The resultant fragment was
cloned into the EcoRI–SalI sites of pUC19, generating the pUC309
plasmid. Competitor DNA fragments of 879 (F879) and 562 bp (F562)
were generated by restriction digestion of pUC309 with BamHI–SspI
and EcoRI–SphI, respectively (Figure 1B). The final competitor used
was the 3 kb Bluescript, pSK1 plasmid (Stratagene). F562 and F879
were damaged by UV irradiation, at 0.1 mW/cm
2
with a germicidal
UV-C lamp. pSK was treated with cisplatin for 15 h in the dark at 37°C
and at a drug:nucleotide ratio of 0.005 (Hansson and Wood,1989).
The CP(–) or CP(1) fragments used in the filter binding assays were
generated by restriction digestion with PvuI of pSK undamaged or
damaged by cisplatin. The AdMLP DNA probe (64mer) was created by
annealing synthesized, complementary oligonucleotides corresponding
to regions –40 to 124 of the AdMLP.
The 32mer 59-TCTTCTTCTTCTTCTGTGCACTCTTCTTCTCT-39
containing a single GpTpG (highlighted sequence) was allowed to react
with cisplatin (Moggs et al., 1996). After ethanol precipitation, the
presence of a 1,3-intrastrand cisplatin d(GpTpG) DNA crosslink was
confirmed by analysis of the oligonucleotide on a 12% acrylamide gel.
The 36 bp DNA probe used in EMSA was created by annealing the
damaged CP(1) or undamaged CP(–) DNA with its complementary
oligonucleotide, leaving a 59 overhang at each extremity. The DNA was
filled and radiolabelled with [
32
P]dATP (3000 Ci/mmol) in the presence
of Klenow and purified on G50 Sephadex columns.
Crude transcription assay
Approximately 15–30 µg of HeLa WCE were incubated with varying
amounts of competitor DNA in a 50 mM Tris–HCl pH 7.9 buffer
containing 10% glycerol, 1 mM EDTA, 0.5 mM dithiothreitol (DTT)
and 5 mM MgCl
2
. Reactions (final volume 20 µl) were incubated for
15 min at 28°C, at which point 50 ng of the AdMLP template (EcoRI–
SalI) were added and pre-initiation of transcription allowed to continue
for 15 min. Transcription was then initiated by addition of NTPs
including [α-
32
P]CTP (400 Ci/mmol). The final volume of the reaction
was 25 µl, and transcription was carried out for 45 min at 28°C. The
RNA transcripts were then analysed by autoradiography and quantified
directly by counting on a PhosphoImage analyser.
The reconstituted transcription assay containing purified transcription
factors TBP, TFIIA, TFIIB, TFIIE, TFIIH, TFIIF and RNA pol II was
modified to include an initial incubation step to allow the potential
binding of transcription factors to a damaged or undamaged fragment
and carried out as described above. TFIID was derived from a subfraction
of the TFIIH purification procedure (Ge
´
rard et al., 1991).
Filter binding assay
Purified recombinant human TBP was combined with various DNA
probes: UV-damaged or undamaged 879 bp fragment was labelled
with [
32
P]dATP (3000 Ci/mmol) using the Klenow fragment of DNA
polymerase; the damaged or undamaged PvuI fragment from plasmid
pSK was labelled by exploiting the exonuclease activity of Klenow in
the presence of [
32
P]dCTP (3000 Ci/mmol) and subsequent filling by
Damaged DNA lures transcription factor TBP/TFIID
addition of cold nucleotides before purification. Approximately 1 ng of
probe corresponding to 5000 c.p.m. was combined with varying amounts
of TBP, in 20 µl of the transcription buffer containing 60 µg/ml bovine
serum albumin (BSA), 500 ng of poly(dGdC) and 5 mM MgCl
2
, for
30 min at 30°C. Reactions were applied to a 0.45 mm nitrocellulose
membrane (Millipore) using the 96-well Hybri-dot Manifold (BRL), pre-
soaked in 0.4 mM KOH, washed with distilled water and pre-equilibrated
in the reaction buffer without BSA. Filters were air dried and directly
exposed to a PhosphoImage screen, for quantification, or a Biomax film
(Kodak). One µl of input DNA corresponding to the same volume used
in each reaction was spotted on Whatman filter paper as a control for
determination of the percentage of DNA retained on nitrocellulose filters.
The amount of radioactivity retained in the presence of TBP was
measured, background counts (radioactivity retained in the absence of
protein) subtracted, and the amount was divided by the level of
radioactivity present in the input.
Miroinjection of rTBP into human fibroblasts
Microneedle injection into homopolykaryons of fibroblasts derived from
a repair-competent individual (C5RO) and of a CS-B patient (CS1AN)
was performed as described earlier (Vermeulen et al., 1994). RNA
synthesis was determined by pulse labelling the cells for 1 h with
[
3
H]uridine (10 µCi/µl), whereas NER was determined by [
3
H]thymidine
(10 µCi/µl) incorporation after UV irradiation (16 J/m
2
) and autoradio-
graphy. TPB and TFIIB were diluted into phosphate-buffered saline
(PBS) containing BSA.
Electrophoretic mobility shift assays
EMSA reaction mixtures (20 µl) contained 0.2 ng of
32
P-labelled 36 bp
DNA probe (10 000 c.p.m.), 500 ng of poly(dGdC) in a 50 mM Tris–
HCl pH 7.9 buffer containing 80 mM KCl, 5 mM MgCl
2
, 0.1 mM
EDTA, 500 ng of BSA, 10% glycerol, 0.5 mM DTT, 0.01% NP-40, and
rTBP and/or rTFIIB, when indicated. After 30 min of incubation at
30°C, glycerol was added to a final concentration of 20% and applied
to a 4% native polyacrylamide gel. Protein–DNA complexes were
electrophoresed in 25 mM Tris–19 mM glycine buffer at room temper-
ature. Gels were dried and exposed to Biomax film (Kodak).
Structural analysis
The crystallographic coordinates of the human TBP–TATA box complex
(Juo et al., 1996) and of a double-stranded DNA dodecamer containing
a central G∧G site (cis-[Pt(NH
3
)
2
-{d(GpG)-N7(G
6
), N7(G
7
)}] intrastrand
crosslink) (Takahara et al., 1995) were extracted from the PDB (Bernstein
et al., 1977; access codes 1TGH and 1GPG respectively). In the platinated
DNA crystal structure, two duplexes are found in the asymmetric unit
but appear to be almost identical (r.m.s.d. value of 0.3 Å between the
two molecules). The structure superpositions were done using the LSQ
options of the program O (Jones et al., 1991). The TBP–DNA interfaces
were analysed and displayed with the program GRASP (Nicholls et al.,
1993). For comparison, the Dickerson’s dodecamer (Drew et al., 1981)
was used as canonical B-DNA (PBD code 1BNA), and noted BDNA.
Acknowledgements
We thank P.Hanawalt, P.Chambon and T.Seroz for fruitful discussion.
We thank M.Chipoulet and A.Fery for their excellent technical assistance
and R.Ripp for help with SETOR. P.V. was supported by NSF and ARC
fellowships. This work was supported by grants from the INSERM, the
CNRS, the Ministe
`
re de la Recherche et de l’Enseignement Supe
´
rieur,
the Association pour la Recherche sur le Cancer and the Direction des
Recherches Etudes et Techniques.
References
Aboussekhra,A.M. et al. (1995) Mammalian DNA nucleotide excision
repair reconstituted with purified protein components. Cell, 80, 859–
868.
Arndt,K.M., Ricupero-Hovasse,S. and Winston,F. (1995) TBP mutants
defective in activated transcription in vivo. EMBO J., 14, 1490–1497.
Bernstein,F.C., Koetzle,T.F., Williams,G.J.B., Meyer,E.F., Brice,M.D.,
Rodgers,J.R., Kennard,O., Shimanouchi,T. and Tasumi,M. (1977) The
Protein Data Bank: a computer-based archival file for macromolecular
structures. J. Mol. Biol., 112, 535–542.
Bohr,V.A., Smith,C.A., Okumoto,D.S. and Hanawalt,P.C. (1985) DNA
repair in an active gene: removal of pyrimidine dimers from the
7455
DHFR gene of CHO cells is much more efficient than in the genome
overall. Cell, 40, 359–369.
Buratowski,S., Hahn,S., Guarente,L. and Sharp,P.A. (1989) Five
intermediate complexes in transcription initiation by RNA polymerase
II. Cell, 56, 549–561.
Calsou,P. and Salles,B. (1994) Properties of damage-dependent DNA
incision by nucleotide excision repair in human cell-free extracts.
Nucleic Acids Res., 22, 4937–4942.
Chasman,D.I., Flaherty,K.M., Sharp,P.A. and Kornberg,R.D. (1993)
Crystal structure of yeast TATA-binding protein and model for
interaction with DNA. Proc. Natl Acad. Sci. USA, 90, 8174–8178.
Colgan,J. and Manley,L. (1992) TFIID can be rate limiting in vivo
for TATA-containing, but not TATA-lacking, RNA polymerase II
promoters. Genes Dev., 6(2), 304–315.
Davison,B.L., Egly,J.-M., Mulvihill,E.R. and Chambon,P. (1983)
Formation of stable preinitiation complexes between eukaryotic class
B transcription factors and promoter sequences. Nature, 301, 680–686.
Donahue,B.A., Yin,S., Taylor,J.S., Reines,D. and Hanawalt,P.C. (1994)
Transcript cleavage by RNA polymerase II arrested by a cyclobutane
pyrimidine dimer in the DNA template. Proc. Natl Acad. Sci. USA,
91, 8502–8506.
Drew,H.R., Wing,R.M., Takano,T., Broka,C., Tanaka,S., Ikatura,K. and
Dickerson,R.E. (1981) Structure of a B-DNA dodecamer: conformation
and dynamics. Proc. Natl Acad. Sci. USA, 78, 2179–2183.
Evans,S.V. (1993) SETOR: hardware lighted three-dimensional solid
model representations of macromolecules. J. Mol. Graphics, 11,
134–138.
Ge
´
rard,M., Fischer,L., Moncollin,V., Chipoulet,J.-M., Chambon,P. and
Egly,J.-M. (1991) Purification and interaction properties of the human
RNA polymerase B(II) general transcription factor BTF2. J. Biol.
Chem., 266, 20940–20945.
Hansson,J., and Wood,R.D. (1989) Repair synthesis by human cell
extracts in DNA damaged by cis- and trans-diammine dichloro
platinum(II). Nucleic Acids Res., 17, 8073–8091.
Hoeijmakers,J.H.J., Egly,J.-M. and Vermeulen,W. (1996) TFIIH: a key
component in multiple DNA transactions. Curr. Opin. Genet. Dev., 6,
26–33.
Humbert,S., van Vuuren,H., Lutz,Y., Hoeijmakers,J.H.J., Egly,J.-M. and
Moncollin,V. (1994) p44 and p34 subunits of the BTF2/TFIIH
transcription factor have homologies with SSL1, a yeast protein
involved in DNA repair. EMBO J., 13, 2393–2398.
Iyer,N., Reagan,M.S., Wu.,K.-J., Canagarajah,B. and Friedberg,E.C.
(1996) Interactions involving the human RNA polymerase II
transcription:nucleotide excision repair complex TFIIH, the nucleotide
excision repair protein XPG, and Cockayne syndrome group B (CSB)
protein. Biochemistry, 35, 2157–2167.
Jones,C.J. and Wood,R.D. (1993) Preferential binding of the xeroderma
pigmentosum group A complementing protein to damaged DNA.
Biochemistry, 32, 12096–12104.
Jones,T.A., Zou,J.Y., Cowan,S.W. and Kjeldgaard,M. (1991) Improved
methods for building protein models in electron density maps and the
location of errors in these models. Acta Crystallogr., A47, 110–119.
Juo,Z.S., Chiu,T.K., Leiberman,P.M., Baikalov,I., Berk,A.J. and
Dickerson,R.E. (1996) How proteins recognize the TATA box. J. Mol.
Biol., 261, 239–254.
Kim,J.L., Nikolov,D.B. and Burley,S.K. (1993) Co-crystal structure of
TBP recognizing the minor groove of a TATA element. Nature, 365,
520–527.
Kim,Y., Geiger,J.H., Hahn,S. and Sigler,P.B. (1993) Crystal structure of
a yeast TBP/TATA-box complex. Nature, 365, 512–520.
Leadon,S.A. and Lawrence,D.A. (1991) Strand-selective repair of DNA
damage in the yeast GAL7 gene requires RNA polymerase II. J. Biol.
Chem., 267, 23175–23182.
Lee,M. and Struhl,K. (1995) Mutations on the DNA-binding surface of
TATA-binding protein can specifically impair the response to acidic
activators in vivo. Mol. Cell. Biol., 15, 5461–5469.
Mayne,L.V. and Lehmann,A.R. (1982) Failure of RNA synthesis to
recover after UV irradiation: an early defect in cells from individuals
with Cockayne’s syndrome and xeroderma pigmentosum. Cancer Res.,
42, 1473–1478.
Mellon,I., Spivak,G. and Hanawalt,P.C. (1987) Selective removal of
transcription-blocking DNA damage from the transcribed strand of
the mammalian DHFR gene. Cell, 51, 241–249.
Moggs,J.G., Yarema,K.J., Essigmann,J.M. and Wood,R.D. (1996)
Analysis of incision sites produced by human cell extracts and
purified proteins during nucleotide excision repair of a 1,3-intrastrand
d(GpTpG)-cisplatin adduct. J. Biol. Chem., 271, 7177–7186.
P.Vichi et al.
Nakajima,N., Horikoshi,M. and Roeder,R.G. (1988) Factors involved in
specific transcription by mammalian RNA polymerase II: purification,
genetic specificity, and TATA box-promoter interactions of TFIID.
Mol. Cell. Biol., 8, 4028–4040.
Nicholls,A. (1993) GRASP: Graphical Representation and Analysis of
Surface Properties. Columbia University, New York.
Nocentini,S., Coin,F., Saijo,M., Tanaka,Y. and Egly,J.-M. (1997) DNA
damage recognition by XPA protein promotes efficient recruitment of
TFIIH. J. Biol. Chem., 272, 22991–22992.
Park,C.H., Mu,D., Reardon,J.T. and Sancar,A. (1995) The general
transcription-repair factor TFIIH is recruited to the excision repair
complex by the XPA protein independent of the TFIIE transcription
factor. J. Biol. Chem., 270, 4896–4902.
Rossignol,M., Kolb-Cheynel,I. and Egly,J.-M. (1997) Substrate
specificity of the cdk-activating kinase (CAK) is altered upon
association with TFIIH. EMBO J., 16, 1628–1637.
Svejstrup,J.C., Vichi,P. and Egly,J.-M. (1996) The multiple roles of
transcription/repair factor TFIIH. Trends Biochem. Sci., 21, 346–350.
Sweder,K.S. and Hanawalt,P.C. (1992) Preferential repair of cyclobutane
pyrimidine dimer in the transcribed strand of a gene in yeast
chromosomes and plasmids is dependent on transcription. Proc. Natl
Acad. Sci. USA, 89, 10696–10700.
Takahara,P.M., Rosenzweig,A.C., Frederick,C.A. and Lippard,S.J. (1995)
Crystal structure of double-stranded DNA containing the major adduct
of the anticancer drug cisplatin. Nature, 377, 649–652.
Treiber,D.K., Zhai,X., Jantzen,H.-M. and Essigman,J.M. (1994)
Cisplatin–DNA adducts are molecular decoys for the ribosomal RNA
transcription factor hUBF (human upstream binding factor). Proc.
Natl Acad. Sci. USA, 91, 5672–5676.
Vassylyev,D.G., Kashiwagi,T., Mikami,Y., Ariyoshi,M., Iwai,S.,
Ohtsuka,E. and Morikawa,K. (1995) Atomic model of a pyrimidine
dimer excision repair enzyme complexed with a DNA substrate:
structural basis for damaged DNA recognition. Cell, 83, 773–782.
Vermeulen,W. et al. (1994) Three unusual repair deficiencies associated
with transcription factor BTF2 (TFIIH). Evidence for the existence of
a transcription syndrome. Cold Spring Harbor Symp. Quant. Biol.,
59, 317–329.
Zamble,D.B. and Lippard,S.J. (1995) Cisplatin and DNA repair in cancer
chemotherapy. Trends Biochem. Sci., 20, 435–439.
Received on July 23, 1997; revised on September 19, 1997
7456
