
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 1 of 24
XAB2 dynamics during DNA damage-
dependent transcriptioninhibition
Lise- Marie Donnio
†
, Elena Cerutti
†
, Charlene Magnani, Damien Neuillet,
Pierre- Olivier Mari, Giuseppina Giglia- Mari*
Institut NeuroMyogène (INMG), CNRS UMR 5310, INSERM U1217, Université Claude
Bernard Lyon 1, Lyon, France
Abstract Xeroderma Pigmentosum group A- binding protein 2 (XAB2) is a multifunctional protein
playing a critical role in distinct cellular processes including transcription, splicing, DNA repair,
and messenger RNA export. In this study, we demonstrate that XAB2 is involved specifically and
exclusively in Transcription- Coupled Nucleotide Excision Repair (TC- NER) reactions and solely for
RNA polymerase 2 (RNAP2)- transcribed genes. Surprisingly, contrary to all the other NER proteins
studied so far, XAB2 does not accumulate on the local UV- C damage; on the contrary, it becomes
more mobile after damage induction. XAB2 mobility is restored when DNA repair reactions are
completed. By scrutinizing from which cellular complex/partner/structure XAB2 is released, we have
identified that XAB2 is detached after DNA damage induction from DNA:RNA hybrids, commonly
known as R- loops, and from the CSA and XPG proteins. This release contributes to the DNA
damage recognition step during TC- NER, as in the absence of XAB2, RNAP2 is blocked longer on
UV lesions. Moreover, we also demonstrate that XAB2 has a role in retaining RNAP2 on its substrate
without any DNA damage.
Editor's evaluation
This manuscript will be of interest for individuals working in genome stability, specifically on the
repair of UV damage and nucleotide excision repair (NER). The authors report that the transcription-
coupled NER factor XAB2 is mobilized after DNA damage and that XAB2 keeps RNA Pol2 engaged
on chromatin. XAB2 mobilization appears to be caused by transcription blockage imposed by the
DNA damage.
Introduction
The DNA molecule in our cells’ nucleus forms the instruction manual for proper cellular functioning.
Unfortunately, the integrity of our DNA is continuously challenged by a variety of endogenous and
exogenous agents (e.g., ultraviolet light [UV], cigarette smoke, environmental pollution, oxidative
damage, etc.). These DNA lesions interfere with DNA replication, transcription, and cell cycle progres-
sion, leading to mutations and cell death, which may cause cancer, inherited diseases, or aging (Chat-
terjee and Walker, 2017).
To prevent the deleterious consequences of persisting DNA lesions, all organisms are equipped
with a network of efficient DNA repair systems. One of these systems is the Nucleotide Excision
Repair (NER) which removes helix- distorting DNA adducts caused by UV such as Cyclo- Pyrimidine
Dimers (CPDs) and 6- 4 Photoproducts (6- 4PPs) (Giglia- Mari etal., 2011).
In mammals, the different steps of NER require more than 30 different proteins that are recruited
sequentially to the DNA damage site, as demonstrated by the different studies of NER proteins
kinetics (Moné etal., 2004; Politi etal., 2005; Rademakers etal., 2003; van den Boom etal., 2004;
RESEARCH ARTICLE
*For correspondence:
†
These authors contributed
equally to this work
Competing interest: The authors
declare that no competing
interests exist.
Funding: See page 22
Preprinted: 24 December 2021
Received: 25 January 2022
Accepted: 25 July 2022
Published: 26 July 2022
Reviewing Editor: Wolf- Dietrich
Heyer, University of California,
Davis, United States
Copyright Donnio, Cerutti
etal. This article is distributed
under the terms of the Creative
Commons Attribution License,
which permits unrestricted use
and redistribution provided that
the original author and source
are credited.

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 2 of 24
Zotter etal., 2006). The first step of NER consists of damage recognition, followed by the opening
of the DNA duplex, dual incisions on both sides of the damage, excision of 24–32 oligonucleotides
containing the damage and, finally, gap- filling by repair DNA synthesis.
NER is divided into two subpathways depending on DNA lesions position within the genome.
The Global Genome repair (GG- NER) will detect and repair lesions throughout the genome, whereas
Transcription- Coupled repair (TC- NER) is associated with RNA polymerase 2 (RNAP2) to repair lesions
on the transcribed strand of active genes (Marteijn etal., 2014).
The NER system has been linked to rare human diseases, classically grouped into three distinct
NER- related syndromes. These include the highly cancer- prone disorder Xeroderma Pigmentosum
(XP) and the two progeroid diseases: Cockayne Syndrome (CS) and Trichothiodystrophy (TTD). Impor-
tantly, CS and TTD patients are not cancer prone but present severe neurological and developmental
features (Hanawalt, 1994).
Xeroderma Pigmentosum group A (XPA)- binding protein 2 (XAB2) is a highly conserved protein
of 100kDa and consists of 15 tetratricopeptide repeat (TPR) motifs that carry out protein–protein
interactions. XAB2 protein was identified as a protein interacting with XPA, a NER factor, using a
yeast two- hybrid system (Nakatsu etal., 2000). Next, it has been shown that XAB2 interacts also
with the TC- NER- specific factors, CSA and CSB, and the elongating form of RNAP2 (Nakatsu etal.,
2000). XAB2 is also essential for early mouse embryogenesis, as demonstrated by the preimplantation
lethality observed in XAB2 knockout mice (Yonemasu etal., 2005).
Downregulation of XAB2, using either anti- XAB2 or siRNA, inhibits normal RNA synthesis and the
recovery of RNA synthesis after UV irradiation (Kuraoka etal., 2008; Nakatsu etal., 2000). Further-
more, injection of anti- XAB2 in GG- NER- deficient cells significantly reduces UV- induced Unscheduled
DNA Synthesis (UDS) during repair (Nakatsu etal., 2000). These results suggest the involvement of
XAB2 in transcription and TC- NER.
Further studies have shown that XAB2 is a component of the Prp19/XAB2 complex (Aquarius [AQR],
XAB2, Prp19, CCDC16, hISY1, and PPIE) or Prp19/CDC5L- related complex required for pre- mRNA
splicing (Kuraoka et al., 2008). XAB2, as well as PRP19 and AQR, has been involved in the DNA
damage response (DDR) (Maréchal etal., 2014; Onyango etal., 2016; Sakasai etal., 2017). Indeed,
XAB2 is essential for homologous recombination (HR) by promoting the end resection step (Onyango
etal., 2016). PRP19 is a sensor of RPA- ssDNA after DNA damage (Maréchal etal., 2014) and AQR
contributes to the maintenance of genomic stability via regulation of HR (Sakasai etal., 2017).
Interestingly, AQR has a role in removing R- loops, a three- stranded nucleic acids structure
composed of a DNA:RNA hybrid and the associated nontemplate single- stranded DNA (Sollier etal.,
2014). These structures can form during transcription, when an RNA molecule emerging from the
transcription machinery hybridizes with its DNA template. They are found abundantly in human gene
promoters and terminators where RNA processing occurs (Wang etal., 2018).
Despite the knowledge acquired in the last decades on XAB2 and its different cellular roles, little
is known about the exact crosstalk and dynamics between its diverse cellular functions, specifically
between DNA repair transcription and splicing. In this work, we describe the molecular dynamics of
XAB2 within the cell after UV- damage induction and during the TC- NER repair process. We determined
in vivo that, in the absence of XAB2, Transcription- Coupled repair reactions are impaired, consequently,
restart of transcription after UV damage is abolished. Surprisingly, unlike all the other NER proteins
studied so far, the mobility of XAB2 is increased after irradiation, and XAB2 shows no accumulation
on local UV lesions. This changing dynamic is not restored until DNA repair is completed. Indeed, in
damaged TC- NER- deficient cells, XAB2 remains more mobile. Interestingly, we demonstrate that, after
DNA damage induction, XAB2 is not released from the splicing complex but is detached from R- loops,
a recently XAB2 identified substrate (Goulielmaki etal., 2021). Additionally, we investigate the rela-
tion between XAB2 and RNAP2, demonstrating that XAB2 retains RNAP2 on its substrate. Moreover, in
the absence of XAB2, RNAP2 interacts strongly and durably with both types of UV lesions (6- 4PPs and
CPDs), suggesting a role of XAB2 in the DNA damage recognition step of TC- NER.
Results
XAB2 is involved in TC-NER process
Two decades ago, Tanaka’s research group demonstrated the involvement of XAB2 in the NER
pathway (Kuraoka etal., 2008; Nakatsu etal., 2000). However, the dynamics of XAB2 during the

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 3 of 24
DNA repair process remained to be elucidated. We aimed to study the shuttling between its different
functions when DNA damage is induced. Firstly, we wanted to verify that XAB2 is exclusively involved
in TC- NER reactions.
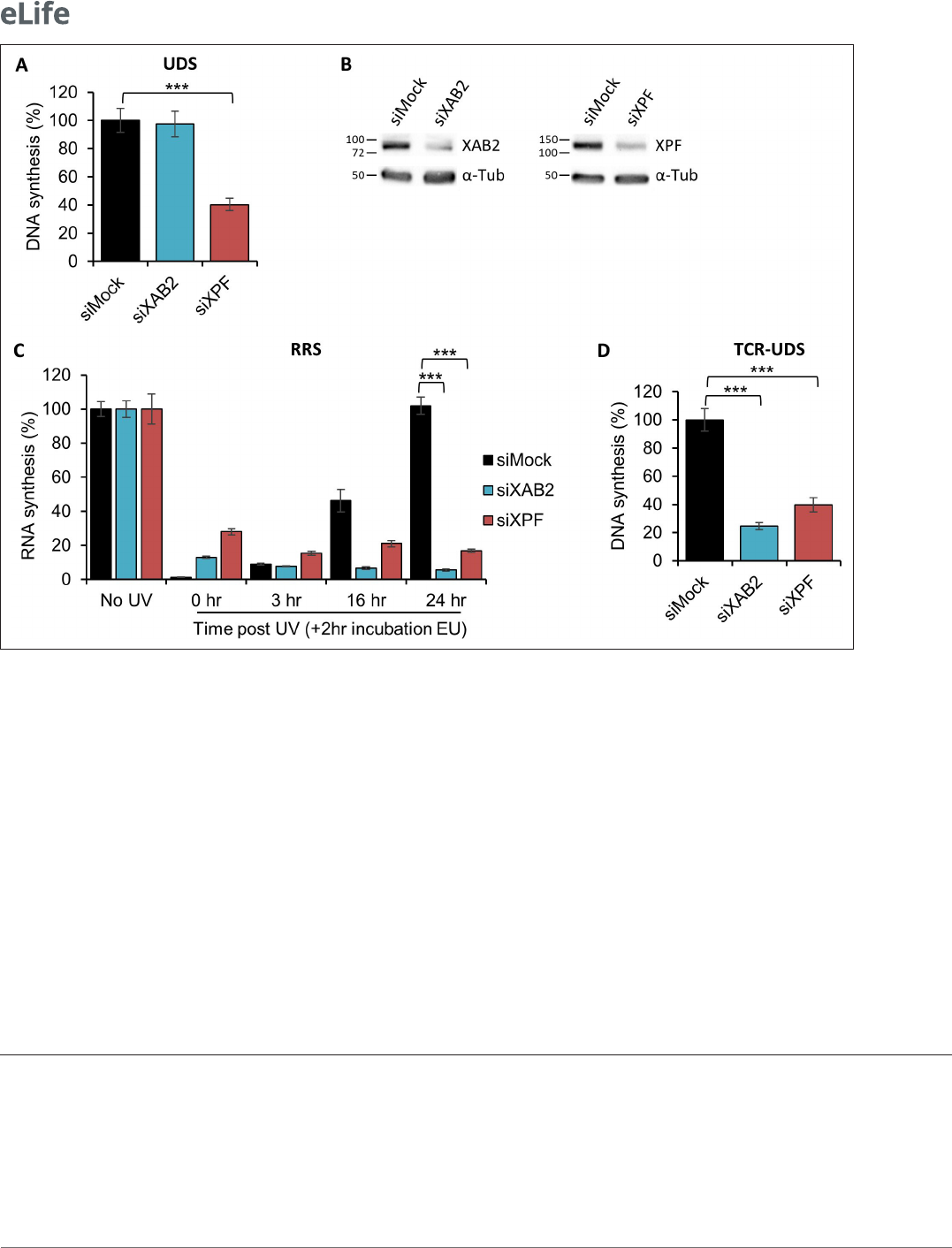
The well- known standard assay used to quantify NER activity is the UDS, which measures repli-
cation activity outside the S- phase after ultraviolet light (UV- C) treatment. This technique quantifies
the refilling of the single- strand DNA gap by the DNA replicative machinery. When we performed
an Unschelduled DNA synthesis (UDS) assay in XAB2- silenced cells, no decreased level of UDS
was observed (as well as in mock- treated cells; Figure1A blue and black columns, Figure1B and
Figure1—figure supplement 1). As a positive control, when we silenced the excision repair factor
XPF, we observed a strong reduction in UDS level (Figure1A, red column, Figure1B, and Figure1—
figure supplement 1 ). This result shows that XAB2 is not involved in the GG- NER subpathway, but
does not exclude an involvement of XAB2 in the TC- NER subpathway.
The commonly used assay measuring TC- NER activity is the RNA Recovery Synthesis (RRS). This
assay measures the newly transcribed RNA by incorporating a nucleoside analog coupled to a fluoro-
phore. The experiment is conducted at different time points after UV irradiation (0, 3, 16, and 24hr)
in order to quantify the decline in transcriptional activity (3hr after UV damage) and the restart of
transcriptional activity (16–24hr after UV irradiation). In XAB2- silenced cells, no restart of transcrip-
tion after UV damage was observed (Figure1C, blue column and Figure1—figure supplement 2A),
as well as in siXPF- treated cells due to the inability to repair DNA lesions (Figure1C, red column
and Figure2—figure supplement 2A) and in contrast with siMock- treated cells (Figure1C, black
column and Figure1—figure supplement 2A). In XAB2- silenced cells and in the absence of DNA
damage, we observed a decreased level of nascent RNA synthesis when the nucleoside analog EU
was incubated for 1 hr, accordingly to the result of Tanaka’s group (Figure1—figure supplement 2B;
Kuraoka etal., 2008; Nakatsu etal., 2000). However, when EU is incubated for 2 hr (time point used
for RRS assay) we observed an increase of nascent RNA in XAB2- silenced cells compared to control
cells (Figure1—figure supplement 2B). As expected, silencing XPF protein does not affect basal
transcription (Figure1—figure supplement 2C). RRS results demonstrated an involvement of XAB2
in the TC- NER subpathway but did not discriminate between a role in the repair reaction per se or in
the Restart of Transcription after Repair (Mourgues etal., 2013).
In order to discriminate this point, we performed an assay designed previously in our group that
precisely measures repair replication during TC- NER: the TCR- UDS assay (Mourgues etal., 2013).
For this assay, we performed the UDS assay in GG- NER- deficient cells using XPC (Xeroderma Pigmen-
tosum complementation group C) mutant cells (XP4PA- SV). The cells were transfected with specific
siRNAs and then locally irradiated with UV- C through a filter. In order to precisely localize DNA-
damaged areas, a γH2AX coimmunofluorescence labeling was performed, and repair replication was
quantified. In siXPF- treated XPC- negative cells, both the GG- NER and the TC- NER pathways are
compromised and, as expected, low TCR- UDS levels were observed compared to siMock- treated
cells (Figure1D, red and black columns and Figure 1—figure supplement 3). Silencing of XAB2
results in a decrease in TCR- UDS levels (Figure1D, blue column and Figure1—figure supplement 3).
This result demonstrates a role of XAB2 in the repair reaction itself, its silencing preventing the DNA
synthesis associated with the excision of UV lesions on actively transcribed genes.
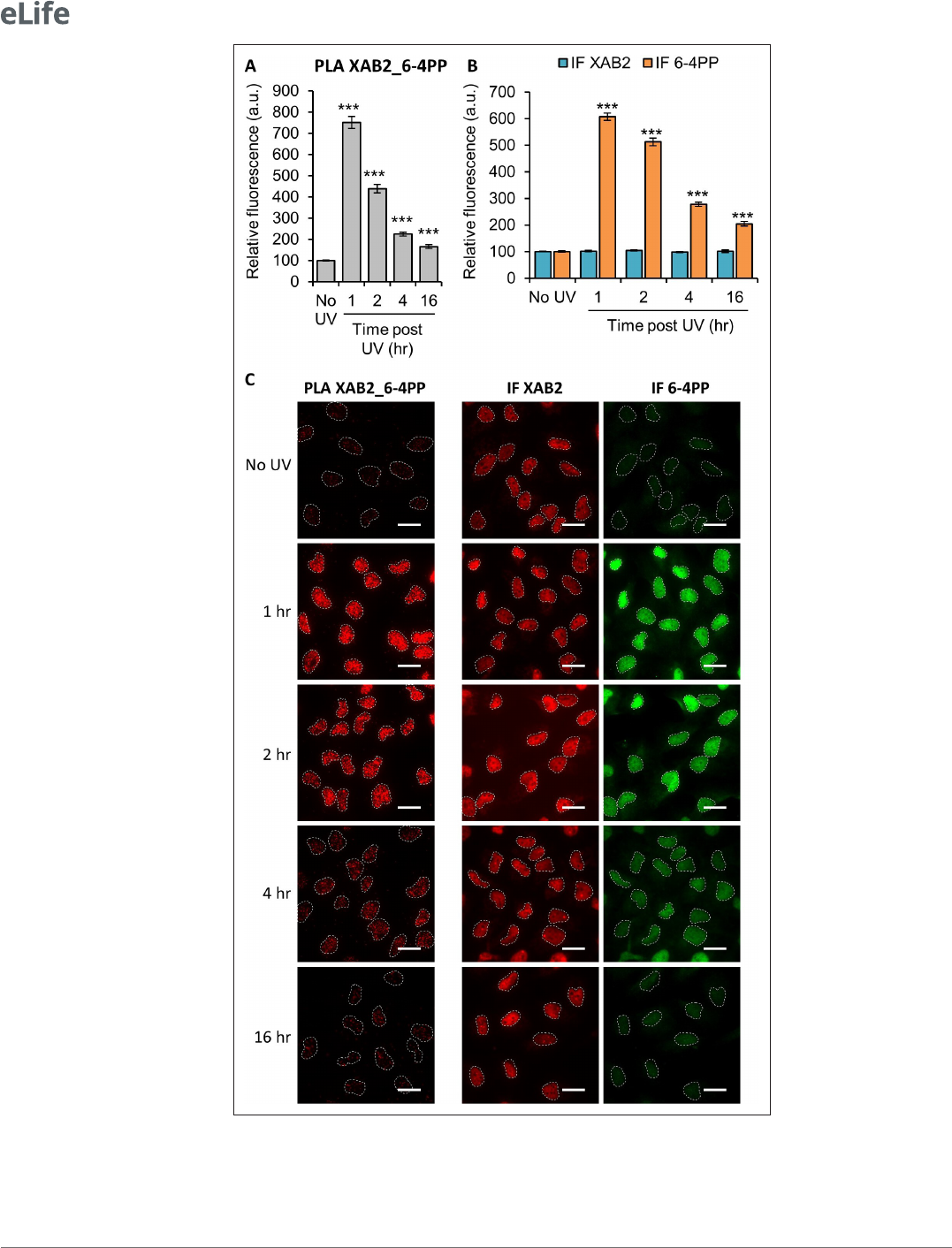
Next, we decided to investigate by PLA (Proximity Ligation Assay) whether XAB2 can interact with
6- 4PPs and CPDs lesion, helix- distorting DNA adducts caused by UV. We observed a strong interac-
tion between XAB2 and 6- 4PPs 1hr after 10J/m² irradiation (Figure2A, C). This interaction correlates
with the amount of 6- 4PPs and, as expected, decreases during repair (Figure 2B, C), while XAB2
concentration does not change after irradiation (Figure2B, C). The same result is obtained in PLA
experiment between XAB2 and CPDs (Figure2—figure supplement 1). These results demonstrate
that XAB2 interacts directly with or is in the proximity of 6- 4PP and CPD lesions until their removal.
Recently, we demonstrate that a fully functional NER mechanism is necessary for the repair of ribo-
somal DNA (rDNA), genes transcribed by the RNA polymerase 1 (RNAP1) (Daniel etal., 2018). To
investigate the involvement of XAB2 in the repair of ribosomal genes, the level of RNAP1 transcription
was measured at different time points after UV irradiation by using a specific ribosomal RNA probe
coupled to a fluorophore, as described previously (Daniel etal., 2018). This probe recognizes the 5′
end of the rDNA transcript, the 47S pre- rRNA (upstream from the first site cleaved rapidly during rRNA
processing) (a sketch of the 47S is depicted in Figure2—figure supplement 2A). In siMock- treated
Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 4 of 24
cells, we observed a decrease of 47S levels 3hr after UV- C exposure and the restart of RNAP1 tran-
scription 40hr after irradiation (Figure2—figure supplement 2B, black column and Figure2—figure
supplement 2D). siCSB- treated cells, deficient for TC- NER, presented a low level of rRNA synthesis
even 40hr after UV- C exposure (Figure2—figure supplement 2B, violet column, Figure2—figure
supplement 2C,D). In the absence of XAB2, 40hr after irradiation, the level of 47S returns to the level
of non- irradiated condition, supporting a restart of RNAP1 transcription (Figure2—figure supple-
ment 2B, blue column and Figure2—figure supplement 2D).
Figure 1. XAB2 is involved in DNA repair. (A) Quantication of Unscheduled DNA Synthesis (UDS) assay determined by EdU incorporation after local
damage (LD) induction with UV- C (100J/m
2
) in WT cells (MRC5 cells) treated with siRNAs against indicated factors. Error bars represent the standard
error of the mean (SEM) obtained from at least 30 LDs. (B) Western blot on whole- cell extracts of MRC5 cells treated with siRNA against indicated
factors. (C) Quantication of RNA Recovery Synthesis (RRS) assay determined by EU incorporation after UV- C (10J/m
2
) exposure in WT cells treated with
siRNAs against indicated factors. Error bars represent the SEM obtained from at least 50 cells. (D) Quantication of TCR- UDS assay determined by EdU
incorporation after LD induction with UV- C (100J/m
2
) in GG- NER- decient cells (XPC−/− cells) treated with siRNAs against indicated factors. Error bars
represent the SEM obtained from at least 15 LDs. For all graphs, p- value of Student’s test compared to siMock condition: ***<0.001.
The online version of this article includes the following source data and gure supplement(s) for gure 1:
Source data 1. Source data for Figure1A: quantication of UDS siXAB2.
Source data 2. Source data for Figure1C: quantication of RRS siXAB2.
Source data 3. Source data for Figure1D: quantication of TCR- UDS siXAB2.
Source data 4. Figures with the uncropped blots and relevant bands clearly labeled for Figure1B: Western blot siXAB2 efciency.
Source data 5. The original les of the full raw unedited gels for Figure1B: Western blot siXAB2 efciency.
Figure supplement 1. UDS siXAB2.
Figure supplement 2. RRS siXAB2.
Figure supplement 2—source data 1. Source data for Figure1—figure supplement 2B, C: quantication of RNA synthesis.
Figure supplement 3. TCR- UDS siXAB2.
Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 5 of 24
Figure 2. XAB2 interacts with the UV lesion 6- 4 Photoproduct (6- 4PP). Quantication of uorescent signal in
the nucleus against the couple XAB2_6- 4PP from Proximity Ligation Assay (PLA) experiment (A) or from the
immunouorescence (IF) done in parallel to PLA assay with the same antibodies dilutions (B). Error bars represent
the standard error of the mean (SEM) obtained from at least 80 cells. P- value of Student’s test compared to No UV
Figure 2 continued on next page

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 6 of 24
All these results demonstrate that XAB2 has a function in TC- NER repair reactions specifically and
exclusively for RNAP2- transcribed genes.
XAB2-splicing complex is released from the DNA damage area
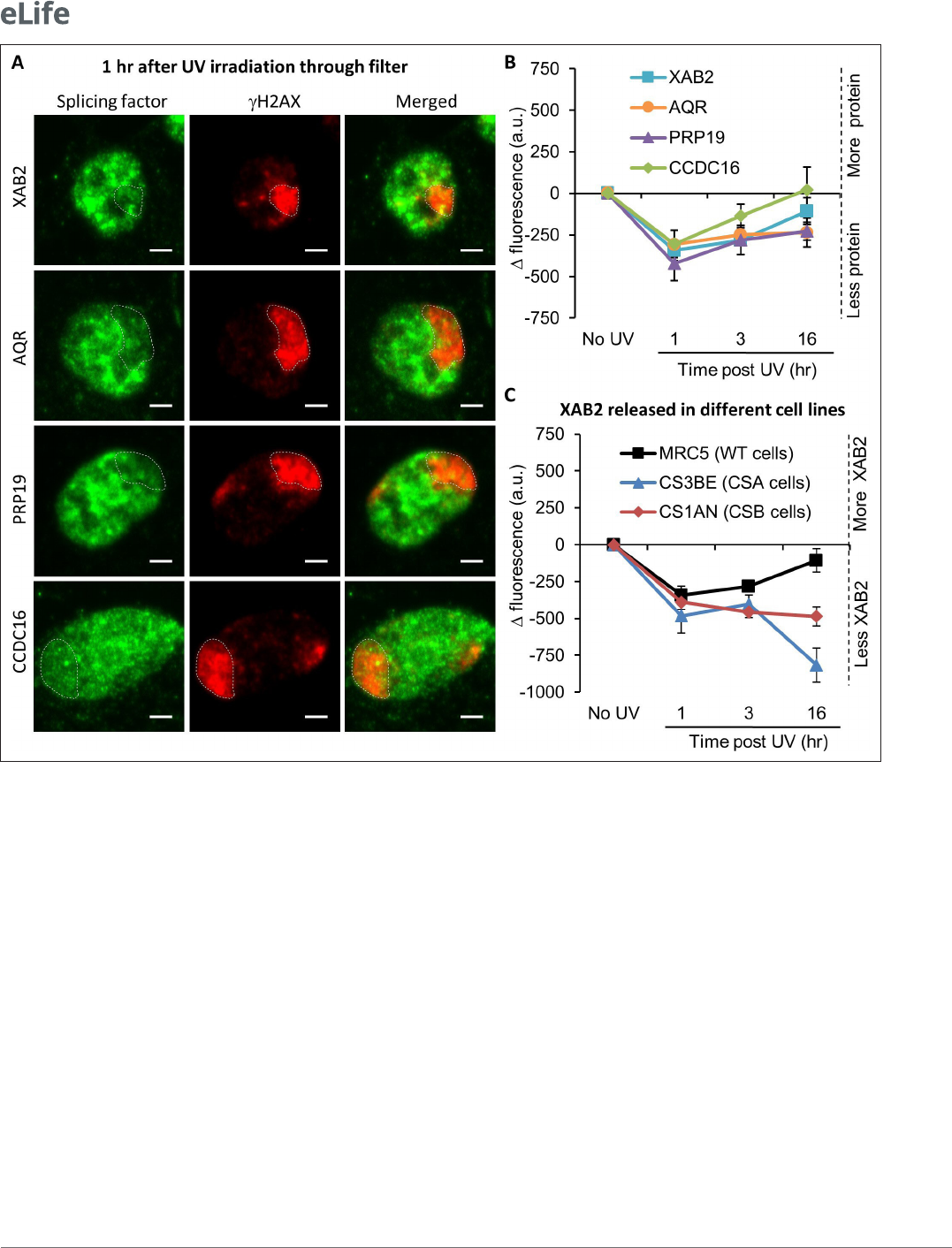
XAB2 is included in a splicing complex composed of five other proteins: Aquarius (AQR), PRP19,
CCDC16, PPIE, and ISY1 (Kuraoka etal., 2008). In order to explore how the XAB2- splicing complex
behaves after local damage induction, the localization of XAB2, AQR, PRP19, and CCDC16 was
revealed by immunofluorescence assays at different time points after local UV irradiation of the cells.
In this assay, the fluorescence signal from each protein in the damaged area (visualized by a costaining
of γH2AX) was compared to the signal from the rest of the nucleus. Unexpectedly, in contrast with all
other NER proteins studied so far, we observed a relatively rapid (1hr after UV irradiation) release of
XAB2, AQR, PRP19, and CCDC16 from the damaged area (Figure3A, B). The proper localization of
the XAB2- splicing complex is re- established after the completion of DNA repair reactions when the
transcription is fully restarted (16hr after irradiation; Figure3B).
We thus verified if the entire XAB2- splicing complex was involved in TC- NER or whether only XAB2
played a role in this process. In order to measure the repair capacity of cells silenced for XAB2- related
proteins, we performed UDS, TCR- UDS, and RRS experiments in AQR/PRP19/CCDC16/PPIE/ISY1-
siRNAs treated cells (Figure3—figure supplements 1–3) and compared the results with XPF- siRNAs
treated cell lines. Our results clearly show that none of the cells silenced for XAB2- related proteins
are deficient in DNA Repair. Moreover, both GG- NER (Figure3—figure supplement 1) and TC- NER
(Figure3—figure supplements 2 and 3) are proficient in the absence of AQR, PRP19, CCDC16, PPIE,
or ISY1.
In order to investigate whether XAB2 release from damaged areas was dependent on the TC- NER
reaction, the localization of XAB2 was detected and quantified within locally damaged areas in
TC- NER- deficient cells: CSA (CS3BE) and CSB (CS1AN) mutant cells. Interestingly, the absence of
CSA and CSB did not hinder the release of XAB2 from locally damaged areas. However, this release
persisted 16hr after UV- C exposure (Figure3C blue and red curves compared to black curve and
Figure3—figure supplement 4), suggesting that the re- establishment of the proper localization of
XAB2 within the nucleus after the DNA repair process depends either on the repair process per se or
on the restart of transcription after the achievement of DNA repair reactions.
XAB2 dynamic during TC-NER
To further analyze XAB2 mobility within the nuclei, we performed SPOT- FRAP (fluorescent recovery
after photobleaching) experiments. In this technique, fluorescence molecules are photobleached in
a small spot by a high- intensity laser pulse. Subsequently, fluorescence recovery within the bleached
area is monitored over time (Figure 4—figure supplement 1A). When cells are untreated, the
measure of fluorescence recovery corresponds to the protein intrinsic mobility within the living cells
(Figure4—figure supplement 1A, black curve). After perturbation of the nuclear environment (e.g.,
condition: ***<0.001. (C) Representative images of the PLA and IF experiments. Nuclei are delimited by dashed
lines. Scale bar: 15µm.
The online version of this article includes the following source data and gure supplement(s) for gure 2:
Source data 1. Source data for Figure2A, B: quantication of PLA and IF XAB2_6- 4PP.
Figure supplement 1. XAB2 interacts with the ultraviolet light (UV) lesions Cyclo- Pyrimidine Dimer (CPD).
Figure supplement 1—source data 1. Source data for Figure2—figure supplement 1A, B: quantication of
PLA and IF XAB2_CPD.
Figure supplement 2. RNA- FISH siXAB2.
Figure supplement 2—source data 1. Source data for Figure2—figure supplement 2B: quantication of RNA-
FISH.
Figure supplement 2—source data 2. Figures with the uncropped blots and the relevant bands clearly labeled
for Figure2—figure supplement 2C: Western blot siCSB efciency.
Figure supplement 2—source data 3. The original les of the full raw unedited gels for Figure2—figure
supplement 2C: Western blot siCSB efciency.
Figure 2 continued
Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 7 of 24
Figure 3. Splicing complex is released from DNA damage. (A) Representative confocal images of immunouorescence (IF) against XAB2, AQR, PRP19,
or CCDC16 (green) and γH2AX (red) 1 hr after local damage (LD) induction with UV- C (60J/m
2
). LDs are indicated by dashed lines. Scale bar: 3µm. (B)
Quantication of the IF signal of the different splicing proteins on the LD after different times of recovery. (C) Quantication of XAB2 signal on LD in
different cell lines after different times of recovery. For both graphs, the signal from the local damage has been subtracted from the background of each
cell. Error bars represent the standard error of the mean (SEM) obtained from at least 20 cells.
The online version of this article includes the following source data and gure supplement(s) for gure 3:
Source data 1. Source data for Figure3B: quantication of splicing complex IF.
Source data 2. Source data for Figure3C: quantication of XAB2 IF.
Figure supplement 1. UDS in splicing complex- silenced cell.
Figure supplement 1—source data 1. Source data for Figure3—figure supplement 1A: quantication of UDS in splicing complex- silenced cells.
Figure supplement 1—source data 2. Figures with the uncropped blots and relevant bands clearly labeled for Figure3—figure supplement 1C:
Western blot efciency of siRNA against splicing complex.
Figure supplement 1—source data 3. The orignal les of the full raw unedited gels for for Figure3—figure supplement 1C: Western blot efciency
of siRNA against splicing complex.
Figure supplement 2. TCR- UDS in splicing complex- silenced cells.
Figure supplement 2—source data 1. Source data for Figure3—figure supplement 2A: quantication of TCR- UDS in splicing complex- silenced
cells.
Figure supplement 3. RRS in splicing complex- silenced cells.
Figure supplement 3—source data 1. Source data for Figure3—figure supplement 3A: quantication of RRS in splicing complex- silenced cells.
Figure supplement 4. XAB2 is released from DNA damage also in TC- NER- decient cells.

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 8 of 24
DNA damage), a protein can physically interacts with a new substrate or a slower complex, becoming
less mobile (Figure4—figure supplement 1A, green curve) or on the contrary can be released from
its substrate, becoming more mobile (Figure4—figure supplement 1A, blue curve). Eventually, the
protein can also have an unchanged mobility (Figure4—figure supplement 1A, red curve).
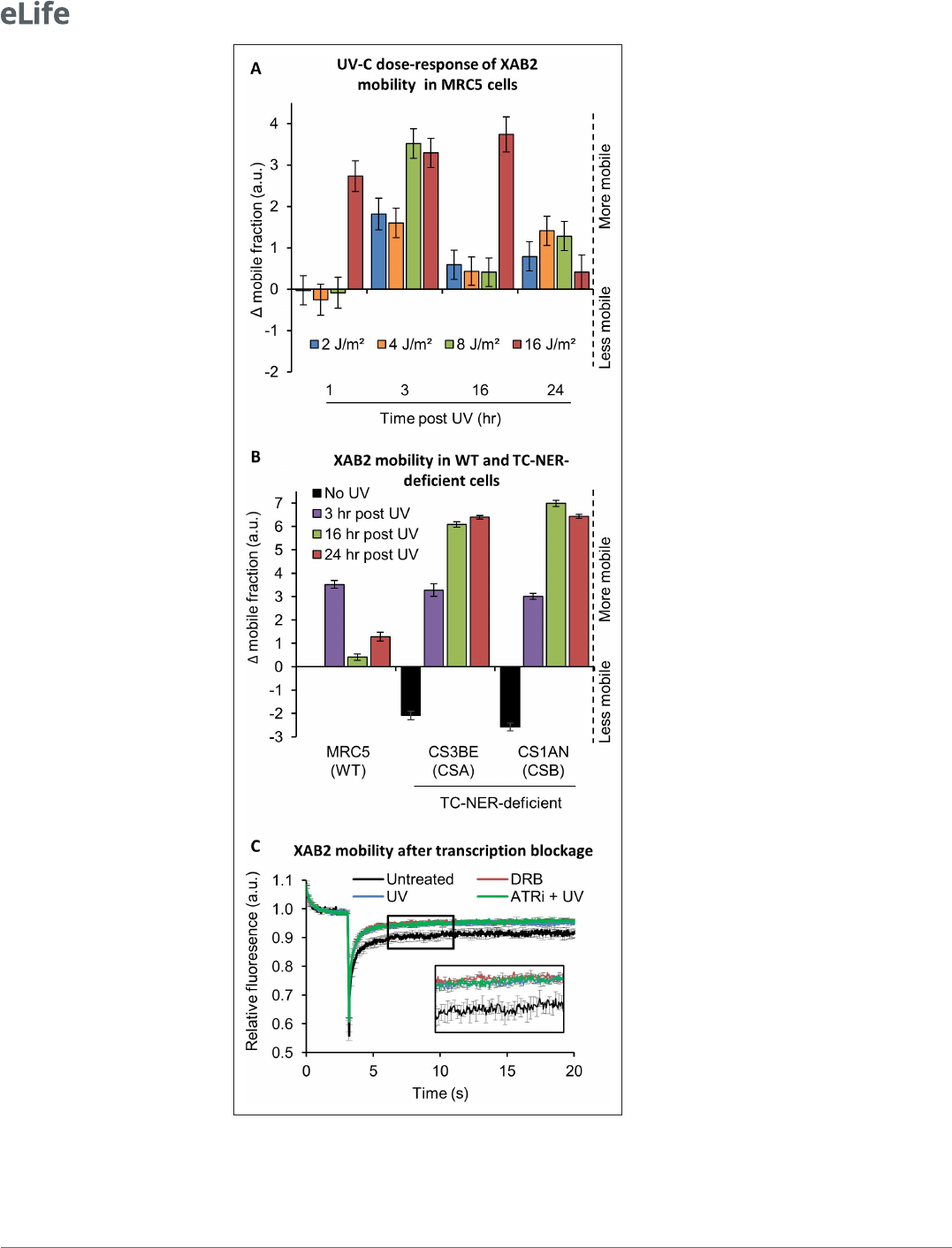
We stably transfected a vector expressing a fluorescent version of XAB2 (XAB2- GFP, Figure4—
figure supplement 1B) in different SV40- immortalized human fibroblast: wild- type cells (MRC5,
Figure4—figure supplement 1C), CSA- deficient cells (CS3BE) and CSB- deficient cells (CS1AN). In
order to determine the minimum dose of UV- C needed to detect a significant difference in XAB2
mobility, MRC5 XAB2- GFP cells were irradiated with doses of UV- C ranging from 2 to 16J/m² and
SPOT- FRAP experiments were performed at different time points following UV irradiation (Figure4A).
Interestingly, we observed a dose- dependent increase in mobility of XAB2 (Figure4A). Doses of UV- C
as weak as 2 and 4J/m² induced a moderate increase in mobility 3 hr post- irradiation and a recovery
of the basal XAB2 mobility within 16 hr post- irradiation (Figure4A, blue and yellow bars). High UV- C
doses (16J/m²) induced a rapid increase in XAB2 mobility (1 hr post- irradiation) and a recovery of
the intrinsic mobility 24 hr post- irradiation (Figure 4A, red bar). At intermediate doses of 8 J/m²
of UV- C, we observed a significant increase in XAB2 mobility during repair (3hr after UV- C expo-
sure) and the following return to the normal condition once the repair is completed and transcription
restarted (16hr after irradiation) (Figure4A, green bar). Interestingly, in CSA and CSB mutant cells,
the increase in XAB2 mobility is also observed after 8J/m² irradiation and lasted until 24 hr after
UV- C exposure (Figure4B), witnessing the fact that in these cells, DNA damage is not repaired and
therefore initial intrinsic XAB2 mobility is not restored. Interestingly, without damage, XAB2 mobility
is reduced in TC- NER- deficient cells compared to wild- type cells for still unknown reasons (Figure4B,
black histogram).
The results of these experiments directed us to explore the possibility that the change in XAB2
mobility was due to transcription inhibition and not really to the repair process itself. In order to verify
this hypothesis, XAB2 mobility was measured after DRB (transcription inhibitor) treatment. Surpris-
ingly, results show that XAB2 increased mobility in transcription inhibition conditions is very similar to
the one measured upon UV treatment (Figure4C, red curve compared to blue curve).
As for XAB2, the mobility of the late- stage spliceosomes changes after UV irradiation. This mobi-
lization depends on DDR signaling pathways (Tresini etal., 2015). Key mediators of DDR are the
ATM and ATR kinases, which induce cell cycle arrest and facilitate DNA repair. To demonstrate that
the change in XAB2 mobility is due (or not) to the UV- damage response, we realized the same FRAP
assays in the presence of ATR and ATM inhibitors. Both drugs did not modify the increase of XAB2
mobility after UV irradiation (Figure4C, green curve and Figure4—figure supplement 1D) demon-
strating that variations in XAB2 mobility after DNA damage are triggered and sustained by transcrip-
tional inhibition.
XAB2 is not released from the splicing complex during DNA repair
reactions
The increase of XAB2 mobility after UV- induced transcription inhibition could be explained by either
the release of XAB2 from a bigger complex and/or the release from an immobile (or nearly immobile)
substrate such as the chromatin or a DNA- related substrate.
In order to distinguish between these two possibilities, we firstly investigated whether, after DNA
damage induction, XAB2 dissociates from its splicing partner AQR by immunoprecipitating XAB2 and
AQR (Figure5—figure supplement 1A). Interestingly, XAB2 was immunoprecipitated more strongly
and consistently 1 hr post- irradiation, time that corresponds to the XAB2 mobility increase. At the
same time point, more AQR is also immunoprecipitated. No clear release of XAB2 from AQR was
observed at different time points.
In parallel, we also verified by PLA whether the binding of XAB2 to AQR was modified after UV- C
irradiation (Figure5—figure supplement 1B). Two hours after UV- C exposure, instead of a release
of XAB2 from AQR, we measured a more robust interaction (Figure 5—figure supplement 1B).
However, this stronger interaction could result from increased AQR concentration 1hr after UV irradi-
ation (Figure5—figure supplement 1C).
In conclusion, immunoprecipitation or PLA experiment showed that XAB2 is not released from the
splicing complex during DNA repair reactions.
Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 9 of 24
Figure 4. XAB2 is dynamic during TC- NER. (A) Fluorescent recovery after photobleaching (FRAP) analysis of XAB2-
GFP mobility in WT cells. Cells were treated or not with different doses of UV- C (2–16J/m
2
) and XAB2 mobility was
measured at different time points after UV- C exposure. The No UV condition was used to calculate the change in
bound fraction. (B) FRAP analysis of XAB2- GFP expressed in WT cells (MRC5- SV) and TC- NER- decient cells (CSA
Figure 4 continued on next page

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 10 of 24
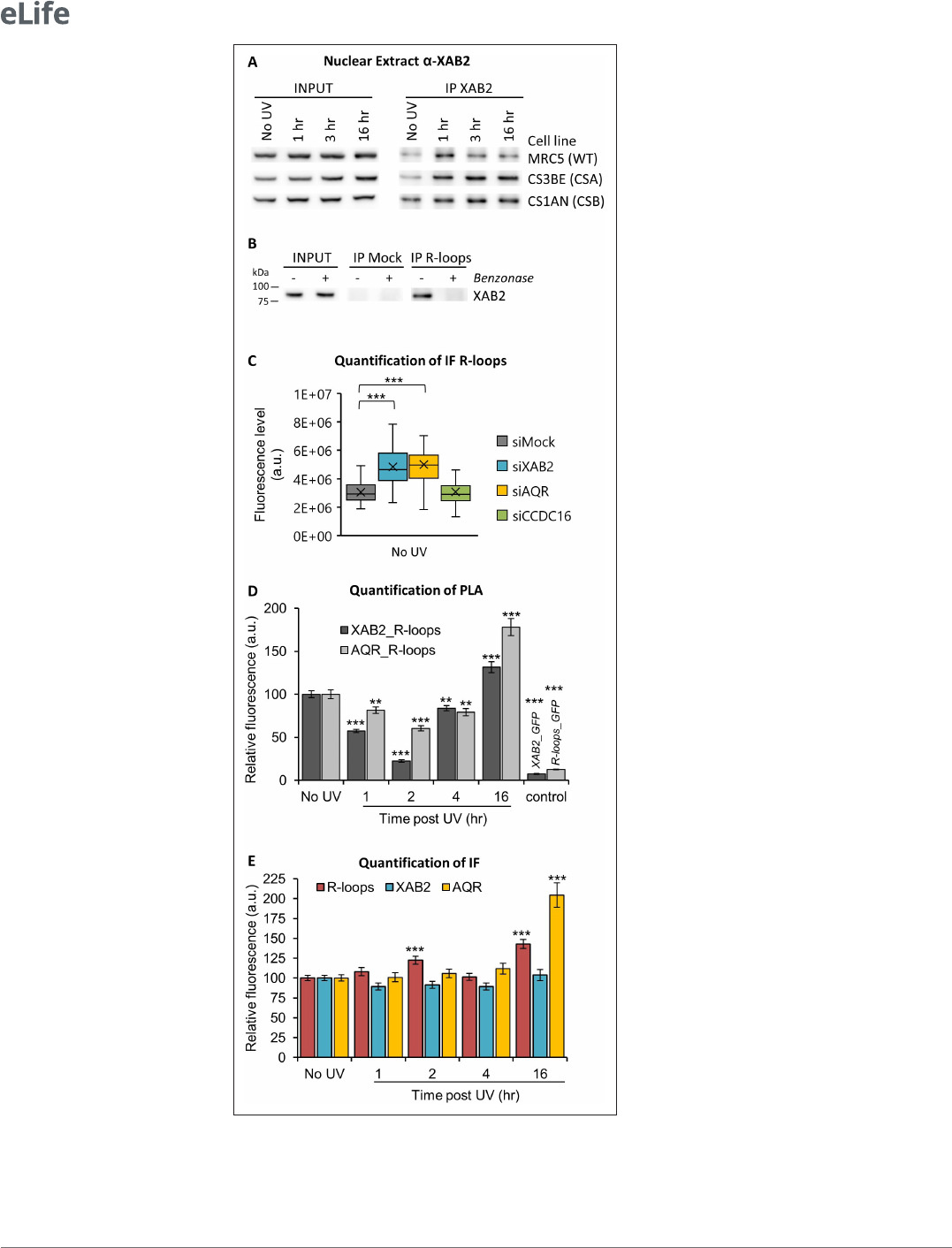
XAB2 is released from R-loops during DNA repair reactions
Interestingly, while trying to immunoprecipitate XAB2 interacting partners during DNA repair reac-
tions, we could observe that systematically and consistently, more XAB2 immunoprecipitated from
nuclear extracts 1–3 hr after UV- C irradiation in WT cells (Figure 5—figure supplement 1A and
Figure5A). At 16 hr post- irradiation, we observed that the amount of XAB2 immunoprecipitated was
comparable to the level observed in non- irradiated cells. Moreover, in CSA−/− and CSB−/− cell lines,
in which UV lesions on transcribed strands of genes are not repaired, the amount of XAB2 immunopre-
cipitated remained high all along the time course of the experiment (3 and 16hr) (Figure5A). These
results hinted that the binding between XAB2 and its substrate is not restored in TCR- deficient cells.
AQR has a role in removing DNA:RNA hybrids, commonly known as R- loops structure (Sollier
et al., 2014) and recently XAB2 was found to be involved in the R- loops resolution (Goulielmaki
etal., 2021). We thus decided to investigate the possible interaction between XAB2 and R- loops.
Immunoprecipitation experiments with the S9.6 antibody show that XAB2 directly interacts with
R- loops (Figure 5B). To test the specificity of the R- loops antibody, nuclear extracts were treated
with Benzonase, an enzyme that specifically degrades DNA:RNA hybrids. Interestingly, after Benzo-
nase treatment, XAB2 is no more immunoprecipitated (Figure5B), suggesting that XAB2 substrate is
indeed DNA:RNA hybrids.
The interaction between XAB2 and R- loops was next investigated by performing IF and PLA exper-
iments with the S9.6 antibody. It has been demonstrated that the S9.6 signal is prominently cyto-
plasmic and nucleolar and derives mainly from ribosomal RNA (Smolka etal., 2021). Consequently,
to increase the specificity of the S9.6 fluorescent signal, the cytoplasm was removed before fixation
(Figure 5—figure supplement 2A) and during quantification, the nucleolar signal was subtracted
from the nuclear signal (Figure5—figure supplement 2B). Using this analysis method, we observed
an increased amount of R- loops in cells silenced for XAB2 or AQR compared to control cells (Figure5C
and Figure5—figure supplement 3A).
Subsequently, we examined whether XAB2 is released from R- loops after DNA damage induc-
tion by performing a PLA assay. After UV irradiation, we measured a strong and consistent reduc-
tion of more than 40% of the interactions between R- loops and XAB2 and between R- loops and
AQR (Figure5D and Figure5—figure supplement 3B). These reduced interactions are not caused
by a reduction in either R- loops, XAB2, or AQR concentration during DNA repair (Figure5E and
Figure5—figure supplement 3B). To verify that this result is specific for R- loops and not coming
from a nonspecific interaction of XAB2 with RNAs, we performed the same assays (PLA and IF) in the
presence of RNAseH which specifically degrades R- loops structures and not single- stranded RNA
(Figure5—figure supplement 4A, B). Our results show that RNAseH reduced the quantity of R- loops
(Figure5—figure supplement 4C,F) and in doing so, it also decreased the interaction with both
XAB2 (Figure5—figure supplement 4C, D) and AQR (Figure5—figure supplement 4E, F). Next,
we verified whether the increase in XAB2 mobility observed after UV irradiation is due to a decreased
interaction with messenger RNA (mRNA) (Figure5—figure supplement 5). PLA results show that
a reduction in the interaction between XAB2 and mRNA was observed (Figure 5—figure supple-
ment 5A, C). However, this reduction paralleled the decrease of mRNA caused by UV- dependent
−/− and CSB−/−). Cells were treated or not with 8J/m
2
of UV- C. The No UV condition of the WT cell lines was
used to calculate the change in bound fraction. (C) FRAP analysis of XAB2- GFP mobility in WT cells after treatment
with 100µg/ml of DRB for 2hr (red line) or with 10J/m
2
of UV- C for 3hr (blue line) or nothing (dark curve). Inhibitor
of ATR pathway was added at 10µM in the medium 1hr before irradiation (green line). For all graphs, error bars
represent the standard error of the mean (SEM) obtained from at least 10 cells.
The online version of this article includes the following source data and gure supplement(s) for gure 4:
Source data 1. Source data for Figure4A: FRAP XAB2- GFP with different doses of UV- C.
Source data 2. Source data for Figure4B: FRAP XAB2- GFP in different cell lines.
Source data 3. Source data for Figure4C: FRAP XAB2- GFP after different treatments.
Figure supplement 1. FRAP of XAB2- GFP.
Figure supplement 1—source data 1. Source data for Figure4—figure supplement 1D: FRAP XAB2- GFP after
different treatments.
Figure 4 continued
Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 11 of 24
Figure 5. XAB2 and AQR are released from R- loops during DNA repair. (A) Immunoprecipitation (IF) of XAB2
in nuclear extract from different cell lines treated with 10J/m
2
of UV- C at different times. Bound proteins were
revealed by Western blotting with antibodies against XAB2. INPUT, 10% of the lysate used for IP reaction. (B) IP
of R- loops in non- crosslinked chromatin extract from WT cells treated or not with Benzonase. XAB2 bounds to
Figure 5 continued on next page

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 12 of 24
transcription inhibition (Figure5—figure supplement 5B, C), suggesting that the results observed in
the PLA XAB2- mRNAs are due mainly to a decrease in mRNAs amount (Figure5—figure supplement
5A).
These results clearly demonstrate that XAB2 is released from R- loops during DNA repair reactions.
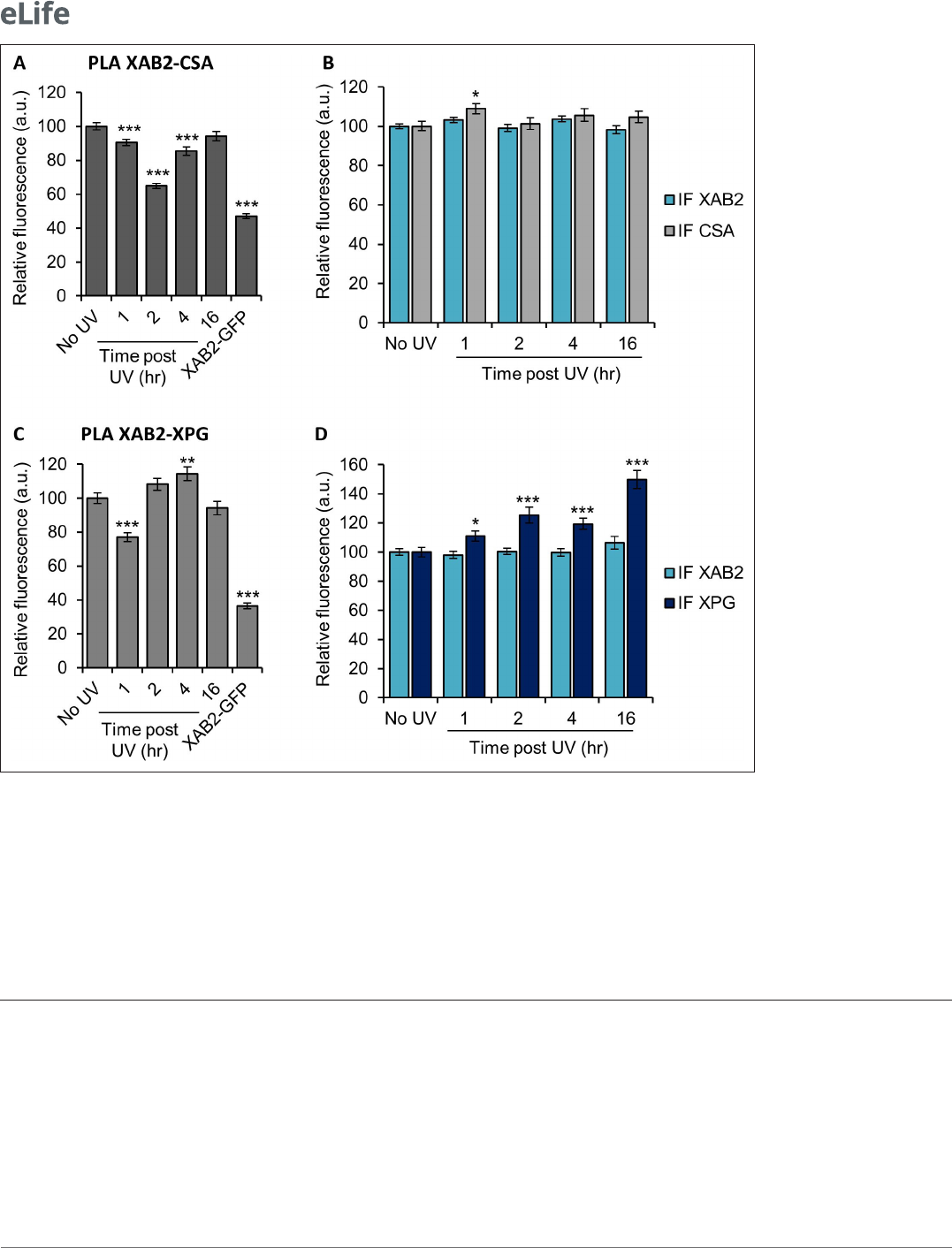
XAB2 is released from CSA and XPG during DNA repair
Because XAB2 has been found to participate specifically in TCR- NER repair reactions (Figure1C), we
wanted to investigate whether part of the increased XAB2 mobility observed after UV induction was
due to a release from repair complexes. We measured, by PLA, the interactions between XAB2 and
CSA, CSB, XPB, or XPG proteins, during TC- NER. Among all the proteins tested, we could observe a
clear and consistent release from the CSA protein 2hr after UV irradiation (Figure6A) and from the
XPG protein 1hr after UV irradiation (Figure6C). The corresponding IF did not show a decreased
quantity of CSA or XPG (Figure6B, D) which validated the specificity of the XAB2- CSA and XAB2- XPG
decreased interactions at those time points. On the contrary, no clear reduction of interaction was
observed between XAB2 and CSB (Figure6—figure supplement 1A, B) or between XAB2 and XPB
(Figure6—figure supplement 1C, D).
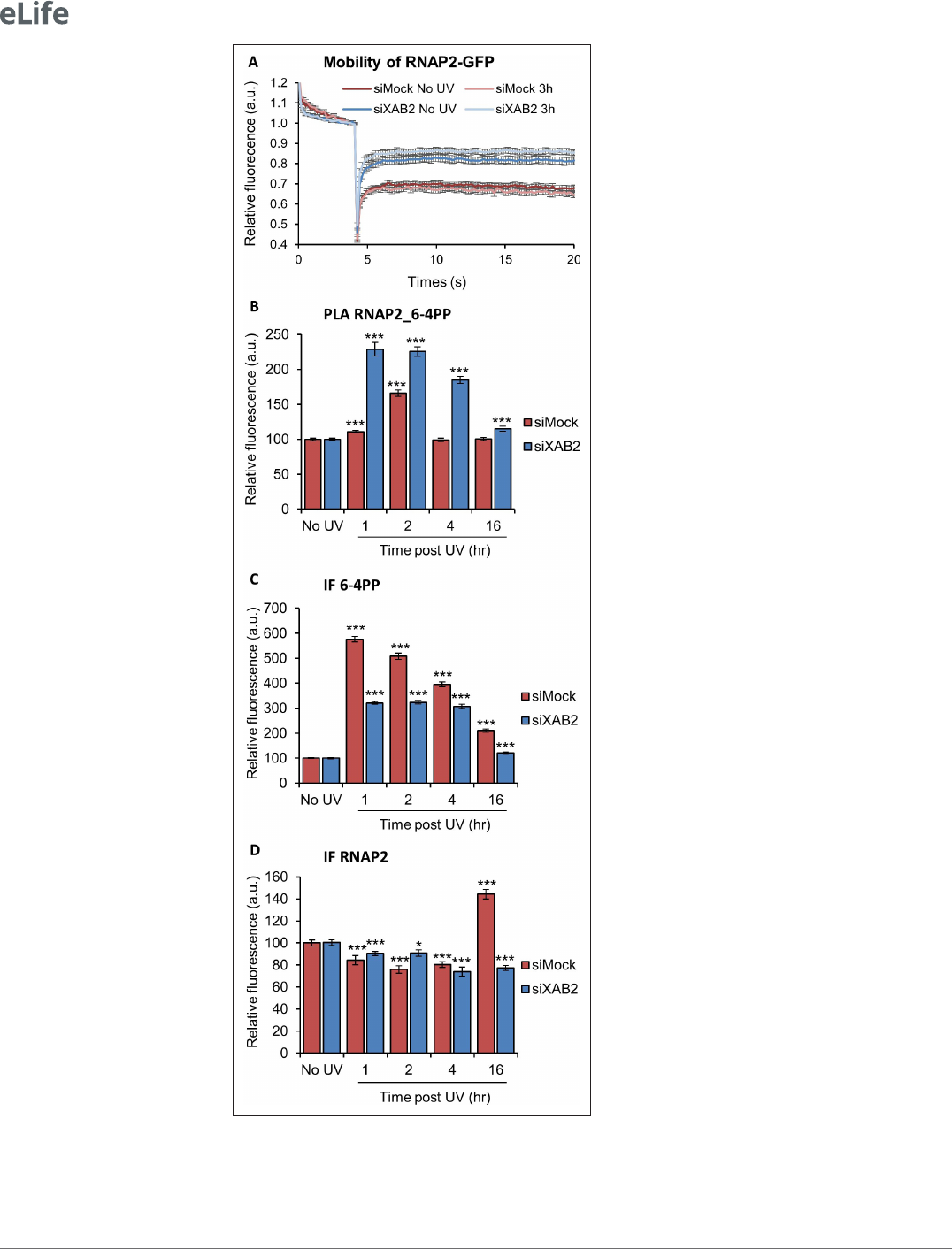
XAB2 depletion modifies RNAP2 behavior
Because XAB2 was found to interact with RNAP2 (Kuraoka etal., 2008; Nakatsu etal., 2000) and
because its increased mobility after irradiation depends on the UV transcription inhibition step, we
R- loops was revealed by Western blotting. INPUT, 10% of the lysate used for IP reaction. (C) Quantication of IF
against R- loops in WT cells treated with siRNAs against indicated factors. Quantication of uorescent signal in
the nucleus against the couple XAB2_R- loops or AQR_R- loops from PLA experiments (D) or from the IF done in
parallel to PLA assays (E). Error bars represent the standard error of the mean (SEM) obtained from at least 50 cells.
P- value of Student’s test compared to No UV or siMock condition: **<0.01; ***<0.001.
The online version of this article includes the following source data and gure supplement(s) for gure 5:
Source data 1. Source data for Figure5C: quantication of IF R- loops in silenced cells.
Source data 2. Source data for Figure5D, E: quantication of PLA and IF XAB2_R- loops and AQR_R- loops.
Source data 3. Figures with the uncropped blots and relevant bands clearly labeled for Figure5A: Western blot
of IP XAB2.
Source data 4. The original les of the full raw unedited gels for Figure5A: Western blot of IP XAB2.
Source data 5. Figures with the uncropped blots and relevant bands clearly labeled for Figure5B: Western blot
of IP R- loops.
Source data 6. The original les of the full raw unedited gels for Figure5B: Western blot of IP R- loops.
Figure supplement 1. Interaction of XAB2 with the splicing complex AQR after UV damage.
Figure supplement 1—source data 1. Source data for Figure5—figure supplement 1B, C: quantication of
PLA and IF XAB2_AQR.
Figure supplement 1—source data 2. Figures with the uncropped blots and relevant bands clearly labeled for
Figure5—figure supplement 1A: Western blot of IP XAB2 in MRC5.
Figure supplement 1—source data 3. The original les of the full raw unedited gels for Figure5—figure
supplement 1A: Western blot of IP XAB2 in MRC5.
Figure supplement 2. IF R- loops and quantication method.
Figure supplement 3. Representatives images of Figure5C,D,E.
Figure supplement 4. Specicity of R- loops antibody.
Figure supplement 4—source data 1. Source data for Figure5—figure supplement 4A: quantication of PLA
and IF R- loops_RNA.
Figure supplement 4—source data 2. Source data for Figure5—figure supplement 4C, E: quantication of
PLA and IF XAB2_R- loops and AQR_R- loops after treatment with RNAseH.
Figure supplement 5. Interaction of XAB2 with RNA.
Figure supplement 5—source data 1. Source data for Figure5—figure supplement 5A,B: quantication of PLA
and IF XAB2_RNA.
Figure 5 continued
Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 13 of 24
wanted to explore whether XAB2 depletion might influence the overall RNAP2 mobility. In order
to investigate this point, we performed FRAP experiments on RNAP2- GFP expressing cells in the
presence or in the absence of XAB2 (Figure 7A; Donnio et al., 2019). Interestingly, depletion of
XAB2 significantly increased the mobility of RNAP2 (Figure7A, dark red curve vs. dark blue curve),
demonstrating that XAB2 maintains RNAP2 bound to its substrate during transcription. Interestingly,
after UV irradiation, RNAP2 mobility did not change significantly (p- value superior at 0.05), both in the
presence and absence of XAB2 (Figure7A, light curve vs. dark curve).
Because FRAP experiments might not reveal more subtle changes in protein–substrate interac-
tions, we decided to examine whether the absence of XAB2 might affect the contacts of RNAP2 with
Figure 6. XAB2 is released from CSA and XPG during DNA repair. Quantication of uorescent signal in the nucleus against the couple XAB2- CSA (A,
B) and XAB2- XPG (C, D) from PLA experiment (A, C) or from the IF done in parallel to PLA assay (B, D). Error bars represent the standard error of the
mean (SEM) obtained from at least 80 cells. P- value of Student’s test compared to No UV condition: *<0.05; **<0.01; ***<0.001.
The online version of this article includes the following source data and gure supplement(s) for gure 6:
Source data 1. Source data for Figure6A, B: quantication of PLA and IF XAB2- CSA.
Source data 2. Source data for Figure6C, D: quantication of PLA and IF XAB2- XPG.
Figure supplement 1. Interaction of XAB2 with CSB or XPB, repair factors of NER.
Figure supplement 1—source data 1. Source data for Figure6—figure supplement 1A, B: quantication of PLA and IF XAB2- CSB.
Figure supplement 1—source data 2. Source data for Figure6—figure supplement 1C, D: quantication of PLA and IF XAB2- XPB.
Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 14 of 24
Figure 7. RNAP2 behavior is modied without XAB2. (A) FRAP analysis of RNAP2- GFP expressing WT cells treated
or not with UV- C (10J/m
2
) after siRNA- mediated knockdown of the indicated factors. Error bars represent the SEM
obtained from at least 10 cells. (B, C, D) Quantication of uorescent signal in the nucleus against the couple
RNAP2_6- 4PP from PLA experiment (B) or from the IFndone in parallel to PLA assay (C, D). Error bars represent
Figure 7 continued on next page

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 15 of 24
UV lesions using the more sensitive PLA assay. Remarkably, we measured a more robust and persistent
interaction of RNAP2 with 6- 4PP lesions after UV irradiation in XAB2- silenced cells (Figure7B and
Figure7—figure supplement 1). The corresponding IF shows a reduced increase of 6- 4PP in XAB2-
silenced cells compared to control cells (Figure7C and Figure7—figure supplement 1) while RNAP2
quantity decreases similarly during DNA repair in the presence or absence of XAB2 due to general
RNAP2 UV- dependent degradation (Figure7D and Figure7—figure supplement 1). These results
demonstrate that, without XAB2, DNA is not properly repaired and as a consequence, RNAP2 inter-
acts longer with UV lesions.
Discussion
Helix- distorting lesions continuously challenge cell survival by interfering with and blocking funda-
mental cellular functions, such as transcription and replication. In order to prevent the deleterious
effects of these events, cells have developed different mechanisms to restore an undamaged DNA
molecule and allow the restart of cellular processes. The importance of rapidly re- establishing
perturbed cellular functions is underlined by the presence of a repair mechanism directly coupled
with transcription, like the TC- NER.
Two phases can be distinguished during TC- NER events: (1) the actual repair reaction of the
damaged strand via the TC- NER subpathway and (2) the resumption of transcription after repair. The
experiment known as ‘RRS’ measures only the restart of transcription after DNA repair completion
and thus the involvement of a protein in the general TC- NER process. However, this assay does not
discriminate between the two phases of the TC- NER. As a consequence, proteins that are fully profi-
cient in the repair reaction but fail in the transcription restart after DNA repair completion might have
the same defect in RRS as those solely deficient in the DNA repair reaction.
Thus far, all studies have demonstrated the involvement of XAB2 in the TC- NER process using only
RRS experiments (Kuraoka etal., 2008; Nakatsu etal., 2000). In this study, we used a specific test
developed in- house (Mourgues etal., 2013) called TCR- UDS and thus demonstrated that, indeed,
XAB2 is solely needed for the repair reaction (Figure 1D). This is confirmed by PLA experiments
showing an interaction between XAB2 and helix- distorting lesions, 6- 4PPs and CPDs, after irradiation
(Figure2 and Figure2—figure supplement 1). However, these experiments do not clearly discrim-
inate at which step of the repair reaction XAB2 may contribute. Two hypotheses are envisaged: (1)
XAB2 functions in damage recognition or (2) in the repair reaction per se.
XAB2 has been found as part of the pre- mRNA splicing complex composed of AQR, PRP19,
CCDC16, PPIE, and ISY1 (Kuraoka et al., 2008). However, none of these proteins take part in
the repair process, underlying the peculiar function of the splicing factor XAB2 in TC- NER repair
(Figure3—figure supplements 1–3).
We also demonstrated that, unlike all the other NER proteins studied so far, XAB2 protein is released
from the damaged region induced by UV- C exposure (Figure3). Concomitantly, we also observed an
increase in XAB2 cellular mobile fraction after UV irradiation (Figure 4A). This release from DNA-
damaged area and increased mobility is surprising and atypical for a repair protein. However, this
behavior has also been observed for late- stage spliceosomes (Tresini et al., 2015) and could be
explained by the importance of the cell rapidly providing access to the repair machinery.
Surprisingly, the increased XAB2 mobility occurs in the absence of CSA and CSB proteins, with
inhibitors of DDR after UV irradiation but also after transcription inhibition (Figure4 and Figure4—
figure supplement 1). These results strongly suggest that XAB2 remobilization is independent of
the repair process but it is more a result of the transcription inhibition induced by the DNA damage.
the standard error of the mean (SEM) obtained from at least 80 cells. P- value of Student’s test compared to No UV
condition: *<0.05; ***<0.001.
The online version of this article includes the following source data and gure supplement(s) for gure 7:
Source data 1. Source data for Figure7A: FRAP RNAP2- GFP.
Source data 2. Source data for Figure7B–D: quantication of PLA and IF RNAP2_6- 4PP.
Figure supplement 1. Representatives images of Figure7B–D.
Figure 7 continued

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 16 of 24
Moreover, the recovery of intrinsic XAB2 mobility is CSA and CSB dependent probably because in
TC- NER defective cells, repair of transcribed genes is deficient and transcription is not recovered. As a
consequence, a proper repair and re- establishment of the transcription process are needed to restore
XAB2 mobility to normal values.
Previously, we reported that a complete TC- NER mechanism is required to repair the UV lesions
present on active rDNA, genes transcribed by RNAP1 (Daniel etal., 2018). Notably, both Cockayne
syndrome proteins (CSA and CSB) are implicated in this specific repair reaction, as well as the UV- stim-
ulated scaffold protein A (UVSSA), a protein required for the stabilization of CSB specifically after
UV irradiation (Higa etal., 2016). By measuring the level of ribosomal RNA, we demonstrate that
RNAP1 transcription restarts after irradiation in the absence of XAB2 (Figure2—figure supplement
2), meaning that UV lesions present on active rDNA are repaired. As a consequence, XAB2 is involved
only in TC- NER of RNAP2- transcribed genes and probably not in the repair of RNAP1- transcribed
genes, confirming a likely specific interaction with RNAP2 and reinforcing the idea that RNAP1 and
RNAP2 repair processes are distinct although they share common proteins.
FRAP experiments in CSA and CSB mutant cells show a more immobile XAB2 fraction than in WT
cells (Figure4B) without any damage induction. Tanaka’s group found that XAB2 interacts in vitro
with CSA and CSB protein in the absence of DNA damage (Nakatsu etal., 2000). In addition, several
studies demonstrated the involvement of CSB in transcription regulation (Boetefuer etal., 2018). As
both CSB and XAB2 are necessary during the transcription process, it is therefore possible that the
absence of CSB will modify XAB2 mobility. However, it is not excluded that in CSA and CSB mutant
cells, a low level of unrepaired oxidative damage (de Waard etal., 2004) might interfere with the
proper XAB2 mobility, eventually modifying the amount of R- loops within these cells. Nevertheless,
it is difficult to precisely estimate the exact number of R- loops between different cell types, and this
hypothesis is difficult to clearly assess.
The UV- induced remobilization of XAB2 is not explained by its release from the splicing complex,
as demonstrated by co- immunoprecipitation and PLA experiments, but it is more the result of the
release from chromatin- specific structures, in this particular case the R- loops (Figure5). An R- loop is
a three- stranded nucleic acid structure composed of a DNA:RNA hybrid and the associated nontem-
plate single- stranded DNA. This structure arises naturally in organisms from bacteria to humans, and
has a multitude of functions in the cell (Belotserkovskii etal., 2018). Our results show that R- loops are
a substrate for XAB2, and after DNA damage induction, the interaction between XAB2 and R- loops is
strongly reduced. This might explain the increased XAB2 mobility during the TC- NER reaction.
It was previously demonstrated that, in AQR- depleted cells, R- loops formation is induced. These
R- loops are actively processed into DNA double- strand breaks by XPF and XPG, the NER endonu-
cleases (Sollier etal., 2014). Without any damage, we also observed an increased level of cellular
R- loops in both AQR- and XAB2- silenced cells (Figure5C), suggesting an involvement of these two
proteins in R- loops resolution, as recently also demonstrated by Goulielmaki etal., 2021.
Transcription process and R- loops formation are finely interconnected. Indeed, R- loops formation
can cause transcription blockage. Transcription blockage due to DNA damage appears to result in
R- loops formation (Mullenders, 2015; Steurer and Marteijn, 2017). Moreover, transcription activity
declines after UV irradiation and mRNAs levels are drastically reduced (Figure5—figure supplement
5). PLA assays show that, after UV irradiation, XAB2 and total RNAs interaction is reduced. However,
because the total amount of RNAs is diminished after transcription block, we assume that XAB2_RNAs
interaction is lessened because of the intrinsic reduced amount of RNAs. Differently from total RNAs,
in our study, we observe that R- loops do not decrease in number after UV damage and transcrip-
tion inhibition (Figure5E). Therefore, the interaction XAB2_R- loops and AQR_R- loops is specifically
hindered after UV damage (Figure5D). However, a careful interpretation of these results should be
made because a recent paper demonstrates that the S9.6 antibody, specific for DNA:RNA hybrids,
can also recognize other nucleic acid structures in immunofluorescence assay (Smolka etal., 2021). To
be able to confirm our results, many additional controls were performed: (1) removal of the cytoplasm
before fixation (Figure5—figure supplement 2A); (2) subtraction of the nucleolar signal from the
nuclear signal (Figure5—figure supplement 2B); (3) treatment with RNAseH which degrade specif-
ically R- loops (Figure5—figure supplement 4); and (4) finally interaction of XAB2 with nascent RNA
(EU staining) is different from interaction with R- loops (Figure5—figure supplement 5). Although all
controls point to a seeming interaction between XAB2 and R- loops or between AQR and R- loops, we
Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 17 of 24
cannot exclude that XAB2 and AQR do not release from DNA:RNA hybrids but from other nucleic acid
structures, for example, double- stranded DNA.
Because XAB2 interacts in vitro with CSA and CSB protein (Nakatsu et al., 2000) and recent
studies have shown an interaction with XPG (Goulielmaki etal., 2021), we decided to verify whether
these interactions were modified during the DNA repair process and the concomitant transcription
inhibition. Our results show a clear and consistent reduction of interaction between XAB2 and CSA
(Figure 6A, B) and between XAB2 and XPG (Figure 6C, D) but not between XAB2 and CSB or
between XAB2 and XPB (Figure6—figure supplement 1). These results suggest that XAB2 inter-
venes in the early steps of TC- NER or at least in the steps that imply the activity of CSB and TFIIH and
most probably the early RNAP2 blocking- recognition step. However, the reduction of interactions
between XAB2 and CSA or XPG is less striking than the one with R- loops. As suggested in Gouliel-
maki etal., 2021, another working hypothesis would be that XAB2 is released from CSA and XPG
proteins associated with R- loops processing.
The physical interaction between XAB2 and RNAP2 has already been established (Kuraoka etal.,
2008; Nakatsu et al., 2000), but the exact relation between XAB2 and RNAP2 has not yet been
disclosed. Without DNA damage, we observed a difference in nascent RNA synthesis in XAB2- silenced
cells compared to control cells (Figure1—figure supplement 2B). Since XAB2 functions in both tran-
scription and splicing, it is not surprising that the quantity of nascent RNA is altered in the absence of
XAB2 (Goulielmaki etal., 2021; Kuraoka etal., 2008). Moreover, we have clearly demonstrated by
FRAP experiments that RNAP2 mobility is severely affected in the absence of XAB2. Namely, in XAB2-
depleted cells, RNAP2 is released from its substrate and its mobility is strongly increased (Figure7A).
RNAP2 immobile fraction after UV irradiation does not change significantly in the presence or absence
of XAB2. However, we could observe that interactions of RNAP2 with the UV lesions (6- 4PPs or CPDs)
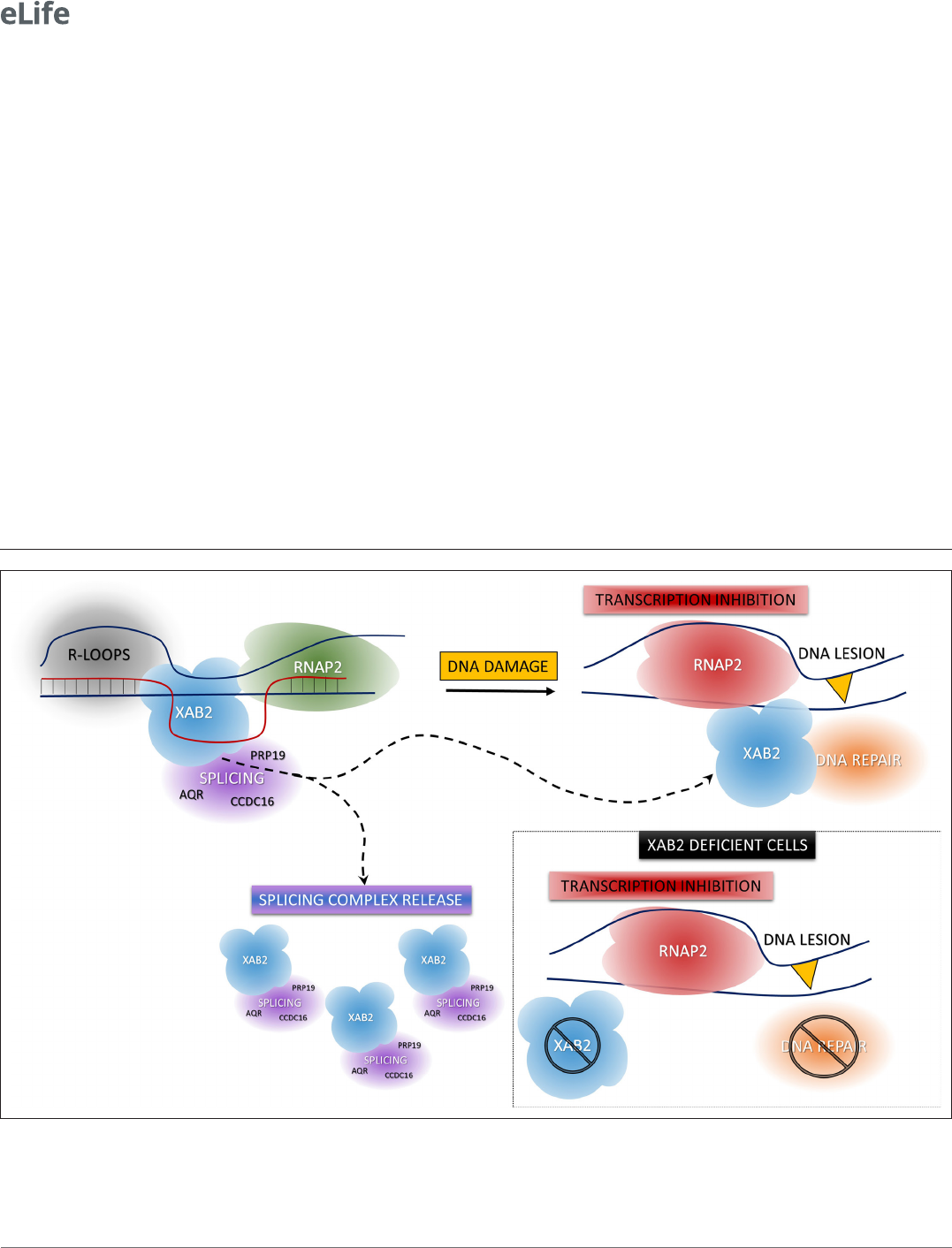
Figure 8. Model of XAB2 dynamics during DNA damage- dependent transcription inhibition. Considering our results, a hypothetical model of XAB2
roles and dynamics can be sketched. XAB2 is involved in R- loops removal and pre- mRNA splicing, both processes linked to transcription. After DNA
damage induction, transcription is blocked and XAB2 (together with some of the proteins involved in the splicing) is massively released from R- loops
allowing a subset of XAB2 molecules to interact with UV- stalled RNA polymerase 2 (RNAP2) and participate in the TC- NER process. In the absence of
XAB2, TC- NER is defective and as a consequence, RNAP2 remains longer in the proximity of DNA lesions.

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 18 of 24
are stronger after DNA damage and last longer in siXAB2- treated cells compared to siMock- treated
cells (Figure7B–D). These results suggest that, during transcription, XAB2 helps RNAP2 anchoring to
its substrate, whereas during the TC- NER process, it has an effect on stalling of RNAP2 on UV lesions,
advocating for a potential role in the DNA damage recognition step.
In conclusion, we describe here an increased mobility of the protein XAB2 during the DNA damage-
dependent transcription inhibition. This increased mobility might partly be explained by the release of
XAB2 from its substrate R- loops and its partner CSA and XPG. Importantly, we demonstrate that XAB2
plays an anchoring role for RNAP2 to its substrate during transcription and helps RNAP2 to detach
from UV lesions after DNA damage. As aconsequence, the absence of XAB2 hinders the overall tran-
scription activity of the cells and severely affects the TC- NER capacity (Figure8).
Materials and methods
Cell culture and treatments
The cells used in this study come from Erasmus MC in Rotterdam and were: (1) wild- type SV40-
immortalized human fibroblasts (MRC5 [RRID:CVCL_D690]); (2) XPC- deficient SV40- immortalized human
fibroblast (XP4PA- SV, GG- NER deficient [RRID:CVCL_6E33]); (3) CSA- deficient SV40- immortalized human
fibroblast (CS3BE, TC- NER deficient [RRID:CVCL_F631]); (4) CSB- deficient SV40- immortalized human
fibroblast (CS1AN, TC- NER deficient [RRID:CVCL_L472]); (5) MRC5- SV stably expressing XAB2- GFP; (6)
CS3BE- SV stably expressing XAB2- GFP; (7) CS1AN- SV stably expressing XAB2- GFP; and (8) MRC5- SV
stably expressing RNAP2- GFP. All cell lines are regularly tested negative for mycoplasma contamination.
Immortalized human fibroblasts were cultured in Dulbecco's Modified Eagle Medium(DMEM from
Sigma) supplemented with 1% of penicillin and streptomycin (Gibco) and 10% fetal bovine serum
(Corning) and incubated at 37°C with 5% CO
2
.
DNA damage was inflicted by UV- C light (254nm, 6- W lamp). Cells were globally irradiated with a
UV- C dose of 2, 4, 8, 10, or 16 J/m
2
or locally irradiated with a UV- C dose of 60 or 100 J/m
2
through
a filter with holes of 5µm of diameter (Millipore). After irradiation, cells were incubated at 37°C with
5% CO
2
for different periods.
Inhibitor of ATR pathway (VE821) and ATM pathway (KU55933) was added at 10µM in the medium
1hr before irradiation.
Construction and expression of RNAP2-GFP and XAB2-GFP fusion
protein
Full- length RNAP2 c- DNA was cloned in- frame into the pEGFP- C1 vector (Clontech), and full- length
XAB2 cDNA was cloned in- frame into the pEGFP- N1 vector. Constructs were sequenced prior to
transfection.
XAB2- GFP and RNAP2- GFP stably expressing cell lines were produced by transfecting XAB2- GFP
or RNAP2- GFP in MRC5, CSA, or CSB cells using FuGENE 6 Transfection Reagent (Promega) according
to the manufacturer’ protocol. The selection was performed with G418 at 2mg/ml.
Transfection of small interfering RNAs
The small interfering RNA (siRNAs) used in this study are: siMock, Horizon, D- 001206- 14 (10 nM);
siXAB2, Horizon, L- 004914- 01 (20nM); siXPF, Horizon, M- 019946- 00 (10nM); siAQR, CCAG ACCA
CUUC CCAU UCU (10nM); siPRP19, GGUG UACA UGGA CAUC AAG (10nM); siCCDC16, GCGA UCUA
GUUU CAUU AAA (5nM); siPPIE, GGC UAUG AGGC AAGU CAAC (5nM); siISY1, GGAA AUCG AGGU
UACA AGU (5nM) and siCSB, Horizon, L- 004888- 00 (10nM). The final concentration used for each
siRNA is indicated in parentheses. All siRNA from Horizon are a pool of four different siRNA.
Cells were seeded in 6- well plates and allowed to attach for at least 24hr. Coverslips were added
inside the well if needed for the experiment. Cells were transfected two times with an interval of 24
hr with siRNA using Lipofectamine RNAiMAX reagent (Invitrogen; 13778150) or GenJet (Tebu- Bio),
according to the manufacturer’s protocol. Experiments were performed between 24 and 72hr after
the second transfection. Protein knockdown was confirmed for each experiment by Western blot.
RRS assay
MRC5 cells were grown on 18mm coverslips. siRNA transfections were performed 24 and 48hr before
the RRS assay. RNA detection was done using a Click- iT RNA Alexa Fluor Imaging kit (Invitrogen),

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 19 of 24
according to the manufacturer’s instructions. Briefly, cells were UV- C irradiated (10J/m²) and incu-
bated for 0, 3, 16, and 24hr at 37°C. Then, cells were incubated for 2hr with 100µM of 5- ethynyl
uridine (EU). After fixation with 4% paraformaldehyde (PFA) for 15min at 37°C and permeabilization
with phosphate- buffered saline (PBS) and 0.5% Triton X- 100 for 20 min, cells were incubated for
30min with the Click- iT reaction cocktail containing Alexa Fluor Azide 594. After washing, the cover-
slips were mounted with Vectashield (Vector). Using ImageJ, the average fluorescence intensity per
nucleus was estimated after background subtraction and normalized to not treated cells.
For RRS with siRNA against splicing protein, cells were incubated for 1 hr with 2.5 mM of
5- bromouracile (BrU). Next, the protocol is the same as immunofluorescence. The primary antibody
used is mouse anti- BrU diluted in PBS+ (PBS containing 0.15 glycine and 0.5% bovine serum albu-
min[BSA]) at 1/750 (11170376001 [sigma]).
RNA fluorescence in situ hybridization
Cells were grown on 18mm coverslips, washed with warm PBS, and fixed with 4% PFA for 15min at
37°C. After two washes with PBS, cells were permeabilized with PBS + 0.4% Triton X- 100 for 7min
at 4°C. Cells were washed rapidly with PBS before incubation (at least 30 min) with prehybridiza-
tion buffer: 15% formamide in 2× SSPE (Sodium Chloride- Sodium Phosphate- EDTA) (0.3 M NaCl,
15.7mM NaH
2
PO
4
·H
2
O, and 2.5mM EDAT [Ethylenediaminetetraacetic acid] et pH8.0). 35ng of the
probe was diluted in 70µl of hybridization mix (2× SSPE, 15% formamide, 10% dextran sulfate and
0.5mg/ml tRNA). Hybridization of the probe was conducted overnight at 37°C in a humidified envi-
ronment. Subsequently, cells were washed twice for 20min with prehybridization buffer and once for
20min with 1× SSPE. After extensive washing with PBS, the coverslips were mounted with Vectashield
containing DAPI (Vector). The probe sequence (5′ to 3′) is Cy5- AGAC GAGA ACGC CTGA CACG CACG
GCAC .
UDS or TCR-UDS
MRC5- SV (WT) or XP4PA- SV (GG- NER- deficient) cells were grown on 18mm coverslips. siRNA trans-
fections were performed 24 and 48hr before UDS assays. After local irradiation at 100 J/m
2
with UV- C
through a 5µm pore polycarbonate membrane filter, cells were incubated for 3 or 8hr (UDS and
TCR- UDS, respectively) with 20µM of EdU (5- ethynyl- 2′-deoxyuridine), fixed with 4% PFA for 15min
at 37° C and permeabilized with PBS and 0.5% Triton X- 100 for 20min. Then, cells were blocked with
PBS+ (PBS, 0.15% glycine and 0.5% BSA) for 30min and subsequently incubated for 1hr at room
temperature (RT) with mouse monoclonal anti-γH2AX antibody (Ser139 [Upstate, clone JBW301])
1:500 diluted in PBS+. After extensive washes with PBS containing 0.5% Triton X- 100, cells were incu-
bated for 45min at RT with secondary antibodies conjugated with Alexa Fluor 594 fluorescent dyes
(Molecular Probes, 1:400 dilution in PBS+). Next, cells were washed several times and then incubated
for 30min with the Click- iT reaction cocktail containing Alexa Fluor Azide 488. After washing, the
coverslips were mounted with Vectashield containing DAPI (Vector). Images were analyzed as follows
using ImageJ and a circle of constant size for all images: (1) the background signal was estimated in
the nucleus (avoiding the damage, nucleoli, and other nonspecific signals) and subtracted, (2) the
locally damaged area was defined by using the γH2AX staining, and (3) the average fluorescence
correlated to the EdU incorporation was then measured and thus an estimation of DNA synthesis after
the repair was obtained.
Immunofluorescence
Cells were plated on 12 or 18mm coverslips to reach 70% confluence on the day of the staining. After
two washes with PBS, cells were fixed with 2% PFA for 15min at 37°C. Cells were permeabilized by
three short washes followed by two washes of 10min with PBS + 0.1% Triton X- 100. Blocking of the
nonspecific signal was performed with PBS+ (PBS, 0.5% BSA, 0.15% glycine) for at least 30min. Then,
coverslips were incubated with primary antibody diluted in PBS+ for 2hr at RT or overnight at 4°C
in a moist chamber. After several washes with PBS + 0.1% Triton X- 100 (three short washes and two
of 10min) and a short wash with PBS+, cells were incubated for 1hr at RT in a moist chamber with a
secondary antibody coupled to a fluorochrome (Goat anti- mouse Alexa Fluor 488 [A11001, Invitrogen]
or 594 [A11005] and Goat anti- rabbit Alexa Fluor 488 [A11008] or 594 [A11012], 1/400 dilution in

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 20 of 24
PBS+). After the same washing procedure with PBS instead of PBS + 0.1% Triton X- 100, coverslips
were finally mounted using Vectashield with DAPI (Vector Laboratories).
Treatment with RNAseH (NEB, M0297L) was performed after blocking with PBS+. RNAseH was
diluted in PBS+ to put 6U per coverslip and incubated for 1hr at 37°C. After washing with PBS+ (one
quick and one of 10min), a primary antibody was added.
For local damage immunofluorescence, the variation of fluorescence in the locally irradiated zone
has been calculated, as for the UDS experiment.
For Immunofluorescence with UV lesions antibodies (6- 4PP and CPD), a step of DNA denaturation
with 0.07M NaOH freshly diluted in PBS for 5min at RT was added after permeabilization. After
several washes with PBS + 0.1% Triton X- 100 (three short washes and two of 10min), cells were incu-
bated with primary antibody.
For RNA detection, the protocol was adapted from Petruk etal., 2016. Briefly, cells were incu-
bated for 2hr with 100µM of EU. After fixation, permeabilization, and blocking, a Biotin tag was
added thanks to a click- it reaction and then recognized by an antibody.
Proximity Ligation Assay
PLA experiments were done using Duolink II secondary antibodies and detection kits (Sigma- Aldrich,
#DUO92002, #DUO92004, and #DUO92008) according to the manufacturer’s instructions. The cells
were fixed and permeabilized with the same procedure as immunofluorescence. After blocking 1hr
at 37°C with the Blocking Solution from the kit, the primary antibodies diluted in Antibody Diluent
was incubated at 4°C overnight. After one quick and three washes of 5 min with PLA buffer A,
Duolink secondary antibodies were added and incubated for 1hr at 37°C. After the same washing
procedure with PLA buffer A, if secondary antibodies were in close proximity (<40nm), they were
ligated together to make a closed circle thanks to the incubation of 30min at 37°C with the Duolink
ligation solution. Then, after the same washing procedure, the DNA is amplified and detected by
fluorescence 594 thanks to the incubation of 100min at 37°C with the Duolink amplification solu-
tion. After washing with PLA buffer B, coverslips were mounted using Vectashield with DAPI (Vector
Laboratories).
Cytostripping
To remove the background generated by some antibodies or EdU incorporation, the cytoplasm of
the cells was removed before fixation. After two washes with cold PBS, cells were incubated on ice
5min with cold cytoskeleton buffer (10mM PIPES [piperazin- N,N'-bis(2- ethanesulfonic acide)] pH 6.8;
100mM NaCl; 300mM sucrose; 3mM MgCl
2
; 1mM EGTA [egtazic acid]; 0.5% Triton X- 100) followed
by 5 min with cold cytostripping buffer (10mM Tris–HCl pH 7.4; 10mM NaCl; 3mM MgCl
2
; 1% Tween
40; 0.5% sodium deoxycholate). After three gentle washes with cold PBS, cells were fixed.
Images acquisition and analysis
For RRS, images of the cells were obtained using an Andor spinning disk: Olympus IX 83 inverted
microscope, equipped with a Yokaga CSU- X1 Spinning disk Unit and BOREALIS technology for homo-
geneous illumination. The acquisition software is IQ3.
For RNAFish, UDS, TCR- UDS, and IF of splicing complex after local damage, images of the cells
were obtained using a Zeiss LSM 780 NLO confocal laser scanning microscope and the following
objective: Plan- Apochromat ×63/1.4 oil DIC (Differential Interference Contrast) M27 or ×40/1.3 oil
DIC. The acquisition software is ZEN.
PLA and IF associated with PLA have been performed on a Zeiss Z1 imager right using a ×40/0.75
dry objective. The acquisition software is Metavue.
Images of the cells for each experiment were obtained with the same microscopy system and
constant acquisition parameters. All images were analyzed with ImageJ software. All experiments
have been performed at least two times and are biological replicates.
Error bars represent the standard error of the mean of the biological replicates. Excel was used for
statistical analysis and plotting of all the numerical data. Statistics were performed using a Student’s
test to compare two different conditions (siMock vs. siRNA X or No UV vs. after irradiation) with the
following parameters: two- tailed distribution and two- sample unequal variance (heteroscedastic).

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 21 of 24
Primary antibodies used for IF and PLA
Primary antibodies used for immunofluorescence and PLA experiments were anti- 6- 4PP (mouse,
NM- DND- 002 [Cosmobio] 1/500 dilution), anti- CPD (mouse, NM- DND- 001 [cosmobio], 1/200 dilu-
tion), anti- XAB2 (mouse, sc- 271037 [Santa Cruz Biotechnology], 1/1000 dilution and rabbit, A303- 638A
[Béthyl], 1/500 dilution), anti- AQR (IPB160 rabbit, A302- 547A [Béthyl], 1/500 dilution), anti- CCDC16
(rabbit, HPA027211 [atlas antibodies], 1/250 dilution), anti- PRP19 (rabbit, ab27699 [abcam], 1/500
dilution), anti- DNA:RNA hybrid clone S9.6 (mouse, MABE1095 [Merck Millipore], 1/100 dilution and
rabbit, Ab01137- 23.0 [Absolute antibody], 1/100 dilution), anti- Biotin (rabbit, ab1227 [abcam] 1/1000
dilution), anti- CSA (rabbit, GTX100145 [genetex], 1/400 dilution), anti- and anti- CSB (mouse, sc398022
[santa- cruz], 1/200 dilution), anti- XPB (rabbit, sc293 [santa- cruz], 1/500 dilution), and anti- XPG (rabbit,
sc84663 [santa- cruz], 1/1000 dilution).
Fluorescence recovery after photobleaching
FRAP experiments were performed on a Zeiss LSM 780 NLO confocal laser scanning microscope
(Zeiss), using a ×40/1.3 oil objective under a controlled environment (37°C, 5% CO
2
). A narrow region
of interest (ROI) centered across the nucleus of a living cell was monitored every 20 ms (1% laser inten-
sity of the 488- nm line of a 25- mW Argon laser) until the fluorescence signal reached a steady state
level (after ≈2s). The same region was then photobleached for 20 ms at 100% laser intensity. Recovery
of fluorescence in the bleached ROI was then monitored (1% laser intensity) every 20 ms for about
20s. Analysis of raw data was performed with the ImageJ software. All FRAP data were normalized to
the average prebleached fluorescence after background removal.
XAB2- GFP SPOT FRAP data were analyzed as follows (Figure4—figure supplement 1). The No
UV condition’s average fluorescence (over all cells) was subtracted from the average fluorescence of
the UV- treated conditions. The obtained difference between the two FRAP curves was then summed
point by point, starting from the bleach up to the following 100 measurements, that is, the area
between the curve of interest and the untreated condition curve.
Protein extraction
For verification of siRNA efficiency, cells were cultured in a 6- well plate. The coverslip needed for
the experiment was displaced before fixation, and cells that remained in the dish were collected.
The extraction of total proteins has been performed using the PIERCE RIPA buffer (Thermo, #89900)
complemented with PIC (Protease Inhibitor Cocktail from ROCHE).
For immunoprecipitation, cells cultured in 10 cm dishes were harvested by scraping, and the
pellet was washed once with PBS supplemented with the PIC. The extraction of nuclear proteins has
been performed using the CelLytic NuCLEAR Extraction kit (Sigma- Aldrich) complemented with PIC.
Protein concentration was determined using the Bradford method. The samples were diluted with
Laemmli buffer (10% glycerol, 5% β-mercaptoethanol, 3% sodium dodecyl sulfate, 100mM Tris–HCl
[pH 6.8], bromophenol blue) and heated at 95°C before loading on a SDS- PAGE (sodium dodecyl
sulfate–polyacrylamide gel electrophoresis).
Coimmunoprecipitation
For coimmunoprecipitation, 10µl of protein G magnetic beads (Bio- adembead, Ademtech) were used
per IP. 1µg of anti- XAB2 antibody (rabbit, A303- 638A, Bethyl) were bound to the beads in PBS with
3% BSA 3% for 2hr at 4°C with rotation. 100µg of nuclear extracts were then incubated with beads–
antibodies complex for 2hr at 4°C with rotation. After two washes at 100mM salt, two at 150mM,
and one wash at 100mM, beads were boiled in 2× Laemmli buffer and eluted samples loaded on a
SDS–PAGE.
RNA/DNA hybrid IP
Non- crosslinked MRC5 cells were lysed in 85mM KCl, 5 mM PIPES (pH 8.0), and 0.5% NP- 40 for
10min on ice. Pelleted nuclei were resuspended in RSB buffer (10 mM Tris–HCl pH 7.5, 200 mM
NaCl, 2.5mM MgCl
2
) with 0.2% sodium deoxycholate, 0.1% SDS, 0.05% sodium lauroyl sarcosinate,
and 0.5% Triton X- 100, and extracts were sonicated for 10min (Diagenode Bioruptor, 60 cycles high
power, 10s ON and 20s OFF). Extracts were then diluted 1:4 in RSB with 0.5% Triton X- 100 (RSB- T)

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 22 of 24
and subjected to IP with the S9.6 antibody overnight at 4°C. RNaseA was added during IP at 0.1ng
RNaseA per microgram genomic DNA. Then protein G dynabeads (Invitrogen) washed with RSB- T
were added and incubated for 3hr. Beads were washed 4× with RSB- T and 2× with RSB; then eluted in
2× Laemmli buffer for 10min at 95°C before loading on SDS–PAGE. When indicated, nuclear extracts
were treated with 0.25U/µl Benzonase (Sigma 70664) for 30min at 37°C before IP.
Western blot
Proteins were separated on a SDS–PAGE composed of bisacrylamide (37:5:1), blotted onto a PVDF
(polyvinylidene difluoride) membrane (0.45μm Millipore). The membrane was blocked in PBS- T (PBS
and 0.1% Tween 20) with 5% milk and then incubated for 2hr at RT or overnight at 4°C with the
following primary antibodies diluted in milk PBS- T: Rabbit anti- XAB2, A303- 638A (Bethyl) 1/1000;
Mouse anti- XPF, MS- 1351- P1 (NeoMarkers) 1/500; Mouse anti-α-Tubulin, T6074 (Sigma- Aldrich)
1/50,000; Rabbit anti- AQR (A302- 547A [Bethyl] 1/2000); Rabbit anti- CCDC16 (A301- 419A [Bethyl]
1/2000), Rabbit anti- PPIE (ab154865 [abcam] 1/1000); Rabbit anti- PRP19 (ab27692 [abcam] 1/1000);
Rabbit anti- ISY1 (ab121523 [abcam] 1/500); Mouse anti- UBF (sc13125 [santa- cruz] 1/500); and Goat
anti- CSB (sc10459 [santa- cruz] 1/100).
Subsequently, the membrane was washed repeatedly with PBS- T and incubated 1hr at RT with
the following secondary antibody diluted 1/5000 in milk PBS- T: Goat anti- rabbit IgG HRP conjugate
(170- 6515; BioRad), Rabbit anti- goat IgG HRP conjugate (172- 1034, BioRad) or Goat anti- mouse IgG
HRP conjugate (170- 6516; BioRad). After the same washing procedure, protein bands were visualized
via chemiluminescence (ECL Enhanced Chemiluminescence; Pierce ECL Western Blotting Substrate)
using the ChemiDoc MP system (BioRad).
Acknowledgement
Additional information
Funding
Funder Grant reference number Author
Agence Nationale de la
Recherche
ANR-14-CE10-0009 Giuseppina Giglia-Mari
Institut National Du Cancer PLBIO17-043 Giuseppina Giglia-Mari
Institut National Du Cancer PLBIO19-126 Giuseppina Giglia-Mari
Ligue Contre le Cancer 218398 Giuseppina Giglia-Mari
Electricité de France 218398 Giuseppina Giglia-Mari
The funders had no role in study design, data collection, and interpretation, or the
decision to submit the work for publication.
Author contributions
Lise- Marie Donnio, Data curation, Formal analysis, Validation, Investigation, Visualization, Method-
ology, Writing – original draft, Writing – review and editing; Elena Cerutti, Conceptualization, Formal
analysis, Validation, Investigation, Methodology, Writing – original draft; Charlene Magnani, Damien
Neuillet, Investigation, Methodology; Pierre- Olivier Mari, Data curation, Software, Supervision,
Writing – review and editing; Giuseppina Giglia- Mari, Conceptualization, Supervision, Funding acqui-
sition, Validation, Writing – original draft, Project administration, Writing – review and editing
Author ORCIDs
Lise- Marie Donnio
http://orcid.org/0000-0002-2414-6034
Elena Cerutti
http://orcid.org/0000-0002-4644-4817
Giuseppina Giglia- Mari
http://orcid.org/0000-0003-2001-1965

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 23 of 24
Decision letter and Author response
Decision letter https://doi.org/10.7554/eLife.77094.sa1
Author response https://doi.org/10.7554/eLife.77094.sa2
Additional files
Supplementary files
• MDAR checklist
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting file.
References
Belotserkovskii BP, Tornaletti S, D’Souza AD, Hanawalt PC. 2018. R- loop generation during transcription:
Formation, processing and cellular outcomes. DNA Repair 71:69–81. DOI: https://doi.org/10.1016/j.dnarep.
2018.08.009, PMID: 30190235
Boetefuer EL, Lake RJ, Fan HY. 2018. Mechanistic insights into the regulation of transcription and transcription-
coupled DNA repair by Cockayne syndrome protein B. Nucleic Acids Research 46:7471–7479. DOI: https://doi.
org/10.1093/nar/gky660, PMID: 30032309
Chatterjee N, Walker GC. 2017. Mechanisms of DNA damage, repair, and mutagenesis. Environmental and
Molecular Mutagenesis 58:235–263. DOI: https://doi.org/10.1002/em.22087, PMID: 28485537
Daniel L, Cerutti E, Donnio LM, Nonnekens J, Carrat C, Zahova S, Mari PO, Giglia- Mari G. 2018. Mechanistic
insights in transcription- coupled nucleotide excision repair of ribosomal DNA. PNAS 115:E6770–E6779. DOI:
https://doi.org/10.1073/pnas.1716581115, PMID: 29967171
de Waard H, de Wit J, Andressoo J- O, van Oostrom CTM, Riis B, Weimann A, Poulsen HE, van Steeg H,
Hoeijmakers JHJ, van der Horst GTJ. 2004. Different effects of CSA and CSB deficiency on sensitivity to
oxidative DNA damage. Molecular and Cellular Biology 24:7941–7948. DOI: https://doi.org/10.1128/MCB.24.
18.7941-7948.2004, PMID: 15340056
Donnio LM, Lagarou A, Sueur G, Mari PO, Giglia- Mari G. 2019. CSB- Dependent Cyclin- dependent kinase 9
degradation and RNA polymerase II phosphorylation during transcription- coupled repair. Molecular and
Cellular Biology 39:e00225- 18. DOI: https://doi.org/10.1128/MCB.00225-18, PMID: 30602496
Giglia- Mari G, Zotter A, Vermeulen W. 2011. DNA damage response. Cold Spring Harbor Perspectives in
Biology 3:a000745. DOI: https://doi.org/10.1101/cshperspect.a000745, PMID: 20980439
Goulielmaki E, Tsekrekou M, Batsiotos N, Ascensão- Ferreira M, Ledaki E, Stratigi K, Chatzinikolaou G, Topalis P,
Kosteas T, Altmüller J, Demmers JA, Barbosa- Morais NL, Garinis GA. 2021. The splicing factor XAB2 interacts
with ERCC1- XPF and XPG for R- loop processing. Nature Communications 12:3153. DOI: https://doi.org/10.
1038/s41467-021-23505-1, PMID: 34039990
Hanawalt PC. 1994. Transcription- coupled repair and human disease. Science 266:1957–1958. DOI: https://doi.
org/10.1126/science.7801121, PMID: 7801121
Higa M, Zhang X, Tanaka K, Saijo M. 2016. Stabilization of ultraviolet (uv)- stimulated scaffold protein a by
interaction with ubiquitin- specific peptidase 7 is essential for transcription- coupled nucleotide excision repair.
The Journal of Biological Chemistry 291:13771–13779. DOI: https://doi.org/10.1074/jbc.M116.724658, PMID:
27129218
Kuraoka I, Ito S, Wada T, Hayashida M, Lee L, Saijo M, Nakatsu Y, Matsumoto M, Matsunaga T, Handa H, Qin J,
Nakatani Y, Tanaka K. 2008. Isolation of XAB2 complex involved in pre- mRNA splicing, transcription, and
transcription- coupled repair. The Journal of Biological Chemistry 283:940–950. DOI: https://doi.org/10.1074/
jbc.M706647200, PMID: 17981804
Maréchal A, Li JM, Ji XY, Wu CS, Yazinski SA, Nguyen HD, Liu S, Jiménez AE, Jin J, Zou L. 2014. PRP19
transforms into a sensor of RPA- ssDNA after DNA damage and drives ATR activation via a ubiquitin- mediated
circuitry. Molecular Cell 53:235–246. DOI: https://doi.org/10.1016/j.molcel.2013.11.002, PMID: 24332808
Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JHJ. 2014. Understanding nucleotide excision repair and its
roles in cancer and ageing. Nature Reviews. Molecular Cell Biology 15:465–481. DOI: https://doi.org/10.1038/
nrm3822, PMID: 24954209
Moné MJ, Bernas T, Dinant C, Goedvree FA, Manders EMM, Volker M, Houtsmuller AB, Hoeijmakers JHJ,
Vermeulen W, van Driel R. 2004. In vivo dynamics of chromatin- associated complex formation in mammalian
nucleotide excision repair. PNAS 101:15933–15937. DOI: https://doi.org/10.1073/pnas.0403664101, PMID:
15520397
Mourgues S, Gautier V, Lagarou A, Bordier C, Mourcet A, Slingerland J, Kaddoum L, Coin F, Vermeulen W,
Gonzales de Peredo A, Monsarrat B, Mari PO, Giglia- Mari G. 2013. ELL, a novel TFIIH partner, is involved in
transcription restart after DNA repair. PNAS 110:17927–17932. DOI: https://doi.org/10.1073/pnas.
1305009110, PMID: 24127601
Mullenders L. 2015. DNA damage mediated transcription arrest: Step back to go forward. DNA Repair
36:28–35. DOI: https://doi.org/10.1016/j.dnarep.2015.09.005, PMID: 26422136

Research article
Cell Biology
Donnio, Cerutti etal. eLife 2022;0:e77094. DOI: https://doi.org/10.7554/eLife.77094 24 of 24
Nakatsu Y, Asahina H, Citterio E, Rademakers S, Vermeulen W, Kamiuchi S, Yeo JP, Khaw MC, Saijo M, Kodo N,
Matsuda T, Hoeijmakers JH, Tanaka K. 2000. XAB2, a novel tetratricopeptide repeat protein involved in
transcription- coupled DNA repair and transcription. The Journal of Biological Chemistry 275:34931–34937.
DOI: https://doi.org/10.1074/jbc.M004936200, PMID: 10944529
Onyango DO, Howard SM, Neherin K, Yanez DA, Stark JM. 2016. Tetratricopeptide repeat factor XAB2
mediates the end resection step of homologous recombination. Nucleic Acids Research 44:5702–5716. DOI:
https://doi.org/10.1093/nar/gkw275, PMID: 27084940
Petruk S, Fenstermaker TK, Black KL, Brock HW, Mazo A. 2016. Detection of RNA- DNA association by a
proximity ligation- based method. Scientific Reports 6:27313. DOI: https://doi.org/10.1038/srep27313, PMID:
27256324
Politi A, Moné MJ, Houtsmuller AB, Hoogstraten D, Vermeulen W, Heinrich R, van Driel R. 2005. Mathematical
modeling of nucleotide excision repair reveals efficiency of sequential assembly strategies. Molecular Cell
19:679–690. DOI: https://doi.org/10.1016/j.molcel.2005.06.036, PMID: 16137623
Rademakers S, Volker M, Hoogstraten D, Nigg AL, Moné MJ, Van Zeeland AA, Hoeijmakers JHJ,
Houtsmuller AB, Vermeulen W. 2003. Xeroderma pigmentosum group A protein loads as A separate factor
onto DNA lesions. Molecular and Cellular Biology 23:5755–5767. DOI: https://doi.org/10.1128/MCB.23.16.
5755-5767.2003, PMID: 12897146
Sakasai R, Isono M, Wakasugi M, Hashimoto M, Sunatani Y, Matsui T, Shibata A, Matsunaga T, Iwabuchi K. 2017.
Aquarius is required for proper CtIP expression and homologous recombination repair. Scientific Reports
7:13808. DOI: https://doi.org/10.1038/s41598-017-13695-4, PMID: 29061988
Smolka JA, Sanz LA, Hartono SR, Chédin F. 2021. Recognition of RNA by the S9.6 antibody creates pervasive
artifacts when imaging RNA:DNA hybrids. The Journal of Cell Biology 220:e202004079. DOI: https://doi.org/
10.1083/jcb.202004079, PMID: 33830170
Sollier J, Stork CT, García- Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. 2014. Transcription- coupled
nucleotide excision repair factors promote R- loop- induced genome instability. Molecular Cell 56:777–785. DOI:
https://doi.org/10.1016/j.molcel.2014.10.020, PMID: 25435140
Steurer B, Marteijn JA. 2017. Traveling rocky roads: The consequences of transcription- blocking DNA lesions on
RNA polymerase II. Journal of Molecular Biology 429:3146–3155. DOI: https://doi.org/10.1016/j.jmb.2016.11.
006, PMID: 27851891
Tresini M, Warmerdam DO, Kolovos P, Snijder L, Vrouwe MG, Demmers JAA, van IJcken WFJ, Grosveld FG,
Medema RH, Hoeijmakers JHJ, Mullenders LHF, Vermeulen W, Marteijn JA. 2015. The core spliceosome as
target and effector of non- canonical ATM signalling. Nature 523:53–58. DOI: https://doi.org/10.1038/
nature14512, PMID: 26106861
van den Boom V, Citterio E, Hoogstraten D, Zotter A, Egly J- M, van Cappellen WA, Hoeijmakers JHJ,
Houtsmuller AB, Vermeulen W. 2004. DNA damage stabilizes interaction of CSB with the transcription
elongation machinery. The Journal of Cell Biology 166:27–36. DOI: https://doi.org/10.1083/jcb.200401056,
PMID: 15226310
Wang IX, Grunseich C, Fox J, Burdick J, Zhu Z, Ravazian N, Hafner M, Cheung VG. 2018. Human proteins that
interact with RNA/DNA hybrids. Genome Research 28:1405–1414. DOI: https://doi.org/10.1101/gr.237362.
118, PMID: 30108179
Yonemasu R, Minami M, Nakatsu Y, Takeuchi M, Kuraoka I, Matsuda Y, Higashi Y, Kondoh H, Tanaka K. 2005.
Disruption of mouse XAB2 gene involved in pre- mRNA splicing, transcription and transcription- coupled DNA
repair results in preimplantation lethality. DNA Repair 4:479–491. DOI: https://doi.org/10.1016/j.dnarep.2004.
12.004, PMID: 15725628
Zotter A, Luijsterburg MS, Warmerdam DO, Ibrahim S, Nigg A, van Cappellen WA, Hoeijmakers JHJ, van Driel R,
Vermeulen W, Houtsmuller AB. 2006. Recruitment of the nucleotide excision repair endonuclease XPG to sites
of UV- induced dna damage depends on functional TFIIH. Molecular and Cellular Biology 26:8868–8879. DOI:
https://doi.org/10.1128/MCB.00695-06, PMID: 17000769
