
Analysis of Petroleum
Hydrocarbons in
Environmental Media
Total Petroleum Hydrocarbon Criteria Working Group Series
Volume 1

Analysis of Petroleum
Hydrocarbons in
Environmental Media

These organizations sponsored or contributed to the
completion of this technical document, prepared by
the TPH Criteria Working Group:
American Petroleum Institute
Association for the Environmental Health of Soils
Association of American Railroads
British Petroleum
Chevron Research and Technology Company
Exxon Biomedical Sciences, Inc.
Retec, Inc.
Shell Development Company
United States Air Force, Air Force Research Laboratory
University of Massachusetts

March 1998
Analysis of Petroleum
Hydrocarbons in
Environmental Media
Total Petroleum Hydrocarbon Criteria Working Group Series
Volume 1
SPONSORED BY:
Association of American Railroads
BP Oil Company
United States Air Force, Armstrong
Laboratory, Occupational Medicine
Division
EDITED BY:
Wade Weisman
Air Force Research Laboratory,
Operational Toxicology Branch

Amherst Scientific Publishers
150 Fearing Street
Amherst, Massachusetts 01002
© 1998 by Amherst Scientific Publishers. All rights reserved.
ISBN 1-884-940-14-5
The material contained in this document was obtained from independent and highly respected sources.
Every attempt has been made to ensure accurate, reliable information, however, the publisher cannot be
held responsible for the information or how the information is applied. Opinions expressed in this book are
those of the Total Petroleum Hydrocarbon Criteria Working Group and do not reflect those of the publisher.
This document was prepared by the Total Petroleum Hydrocarbon Criteria Working Group. Neither the
Working Group nor members of the Working Group:
a. Makes any warranty or representation, expressed or implied, with respect to the accuracy, com-
pleteness, or usefulness of the information contained in this report, or that the use of any appa-
ratus, method, or process disclosed in this report may not infringe privately owned rights; or
b. Assumes any liability with respect to the use of, or for damages resulting from the use of,
any information, apparatus, method, or process disclosed in this report.
This document may be reproduced only in its entirety for unlimited distribution. Every reasonable effort
has been made to give reliable data and information, but neither the TPH Criteria Working Group nor the
contributing individuals or their companies assume any responsibility for the validity of all materials or for
the consequences of their use or misuse.
A portion of the proceeds from the sale of this book will be donated to the Plant-a-Tree Program, a refor-
estation program managed by the U.S. Forest Service.
Printed in the United States of America

CONTENTS
PREFACE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . ix
ACKNOWLEDGMENTS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . xi
1. PURPOSE . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
2. INTRODUCTION . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1
3. OVERVIEW OF TPH . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 2
3.1 Risk Implications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5
4. UNDERSTANDING THE PETROLEUM ANALYTICAL PROCESS:
FROM SAMPLE COLLECTION TO MEASUREMENT . . . . . . . . . . . . . . . . . . . . . . . 6
4.1 Collection And Preservation Of Environmental Samples . . . . . . . . . . . . . . 7
4.2 Sample Extraction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .7
Water Samples . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 12
Soil Samples . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 13
Free Phase Hydrocarbon Samples . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
4.3 Concentration of Sample Extract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 14
4.4 Cleanup of Sample Extract . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 15
4.5 Measurement . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 16
Total Petroleum Hydrocarbon (TPH) Measurement . . . . . . . . . . . . . . . . . 18
Petroleum Group Type Measurement . . . . . . . . . . . . . . . . . . . . . . . . . . . 18
Petroleum Constituent Measurement . . . . . . . . . . . . . . . . . . . . . . . . . . 18
5. TOTAL PETROLEUM HYDROCARBON (TPH) MEASUREMENT:
DETAILED REVIEW OF SELECTED ANALYTICAL METHODS . . . . . . . . . . . . . . . . . 18
5.1 Gas Chromatography (GC) TPH Methods . . . . . . . . . . . . . . . . . . . . . . . . 19
Overview of the Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 19
Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
What Do GC Methods Measure? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 23
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 24
5.2 Infrared Spectroscopy (IR) TPH Methods . . . . . . . . . . . . . . . . . . . . . . . . 27
Overview of the Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 29
v

What Do IR Methods Measure? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 30
5.3 Gravimetric TPH Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
Overview of the Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
What Do Gravimetric Methods Measure? . . . . . . . . . . . . . . . . . . . . . . . . 32
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
5.4 Immunoassay TPH Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
Overview of the Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 33
Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
What Do Immunoassay Methods Measure? . . . . . . . . . . . . . . . . . . . . . . 34
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
6. PETROLEUM GROUP TYPE MEASUREMENT: DETAILED REVIEW
OF SELECTED ANALYTICAL METHODS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 34
6.1 Thin Layer Chromatography (TLC) Group Type Methods . . . . . . . . . . . . . . 34
Overview of the Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
What Do TLC Methods Measure? . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 35
6.2 Immunoassay Group Type Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
Overview of the Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
What Do Immunoassay Methods Measure? . . . . . . . . . . . . . . . . . . . . . . 37
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 37
7. INDIVIDUAL PETROLEUM CONSTITUENT MEASUREMENT:
DETAILED REVIEW OF SELECTED ANALYTICAL METHODS . . . . . . . . . . . . . . . . . 38
7.1 Gas Chromatography with Photoionization Detection
(GC/PID) Petroleum Constituent Methods . . . . . . . . . . . . . . . . . . . . . . . . 41
Overview of Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
What Do GC/PID Methods Measure? . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 41
7.2 Gas Chromatography With Flame Ionization Detection
(GC/FID) Petroleum Constituent Methods . . . . . . . . . . . . . . . . . . . . . . . . 42
Overview of Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
vi

Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 42
What Do GC/FID Methods Measure? . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
7.3 High Performance Liquid Chromatography (HPLC) Petroleum
Constituent Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
Overview of Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
What Do HPLC Methods Measure? . . . . . . . . . . . . . . . . . . . . . . . . . . . . 44
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
7.4 Gas Chromatography With Mass Spectrometry Detection
(GC/MS) Petroleum Constituent Methods . . . . . . . . . . . . . . . . . . . . . . . . 45
Overview of Technique . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 45
Example Methods . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
What Do GC/MS Methods Measure? . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
Interferences/Limitations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
8. EVOLVING METHODS FOR PETROLEUM HYDROCARBON FRACTIONS . . . . . . . . . 48
8.1 What Do Petroleum Fraction Methods Measure? . . . . . . . . . . . . . . . . . . 48
8.2 Why Use Petroleum Fraction Methods? . . . . . . . . . . . . . . . . . . . . . . . . . 48
8.3 Examples Of Petroleum Fraction Methods . . . . . . . . . . . . . . . . . . . . . . . 49
TPHCWG Analytical Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
Massachusetts EPH/VPH Method . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
Appendix I - HYDROCARBON CHEMISTRY . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
Appendix II - CHARACTERIZATION OF PETROLEUM PRODUCTS . . . . . . . . . . . . . . . 59
Appendix II-A: PRODUCT COMPOSITION AND SPECIFICATION . . . . . . . . . . . . . . . . 61
Appendix II-B: ENVIRONMENTAL FATE OF PETROLEUM PRODUCTS:
WEATHERING AND TRANSPORT . . . . . . . . . . . . . . . . . . . . . . . . . . 70
Appendix III - Refinery Flow Diagrams . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75
Appendix IV - Quick Reference of TPH Methods . . . . . . . . . . . . . . . . . . . . . . . . . 81
EXPLANATION OF ACRONYMS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
GLOSSARY OF TERMS . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 89
BIBLIOGRAPHY . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 97
vii

PREFACE
This document is the first in a series from the Total Petroleum Hydrocarbon
Criteria Working Group (TPHCWG, or “Working Group”). The Working Group
convened in 1993 to address the large disparity among cleanup requirements
being used by states at sites contaminated with hydrocarbon materials such as fuels,
lubricating oils and crude oils. These requirements usually focus on total petrole-
um hydrocarbon (TPH), with numerical standards ranging from tens to tens of
thousands of milligrams of TPH per kilogram of soil. Recognizing that these stan-
dards are not based on a scientific assessment of human health risk, Working
Group members established the following goal for their effort:
To develop scientifically defensible information for establishing soil cleanup levels that
are protective of human health at petroleum contaminated sites.
The Working Group is guided by a steering committee consisting of representa-
tives from industry, government, and academia. Some of the active participants
among the more than 400 involved include the Gas Research Institute, several
major petroleum companies including Chevron, Exxon, British Petroleum and
Shell, the American Petroleum Institute, the Association of American Railroads,
several state governments (Washington, Texas, Colorado, Hawaii, Louisiana, New
Mexico, Massachusetts), the U.S. Environmental Protection Agency, the
Department of Defense, the University of Massachusetts, and private consulting
firms including EA Engineering, Science & Technology and Menzie-Cura &
Associates, Inc.
The Working Group compiled their data collection and analytical efforts into
five volumes:
Volume 1. Analysis of Petroleum Hydrocarbons in Environmental Media (this
volume) discusses and critiques analytical methods for quantifying
TPH, petroleum mixtures and individual petroleum constituents
in soil and water samples. It is designed to be a reference tool for
the nonchemist, describing what information analytical methods
can provide for risk assessment.
Volume 2. Composition of Petroleum Mixtures (in press) provides the best
available composition information for a variety of petroleum
products.
Volume 3. Selection of Representative Total Petroleum Hydrocarbon (TPH) Fractions
Based on Fate and Transport Considerations (1997, Amherst Scientific
Publishers) defines fractions of TPH expected to behave similarly
in the environment. Identification of these fractions simplifies
analysis of environmental samples, fate and transport modeling,
and risk assessment efforts at petroleum contaminated sites.
ix

Volume 4. Development of Fraction-Specific Reference Doses (RfDs) and Reference
Concentrations (RfCs) for Total Petroleum Hydrocarbons (TPH) (1997,
Amherst Scientific Publishers) provides the technical basis for the
development of TPH fraction-specific RfDs and RfCs for use in the
hazard assessment step of the Working Group’s risk-based
approach to establishing soil cleanup levels at petroleum contami-
nated sites.
Volume 5. Human Health Risk-Based Evaluation Of Petroleum Contaminated Sites:
Implementation Of The Working Group Approach.(in press). This docu-
ment integrates the findings of Volumes 1 through 4 into a risk-
based framework for development of cleanup goals at petroleum
contaminated sites. It includes descriptions of demonstration sites
where the Working Group approach has been used successfully.
Amherst Scientific Publishers will publish these volumes in 1997 and 1998. In
addition to these volumes, results of projects where use of the Working Group
approach has been demonstrated (demonstration sites) and a concise technical
summary document are now or will soon be available on the U.S. Air Force
Toxicology Division web site (http://voyager.wpafb.af.mil). At this web site,
Working Group publications may be downloaded from the “recent publications”
icon. Additional Working Group resources will be added to this web site as they
become available.
We hope you find these documents to be useful in your effort to evaluate and
determine acceptable risk-based cleanup criteria at petroleum contaminated sites.
Wade H. Weisman
Chairman, TPH Criteria Working Group
x

ACKNOWLEDGMENTS
The TPH Criteria Working Group would like to especially thank BP Oil for their
strong support of this effort through permitting the use of the “TPH in Soil Primer”,
developed by Elaine Schwerko for BP Oil, Environmental Technology Branch,
© September, 1993. The development and production of this document was sup-
ported, in part, through the financial contribution of the Association of American
Railroads (AAR). The continued support of TPH Working Group efforts by Dr.
Christopher Barkan of the Environmental and Hazardous Materials Research
Program of the AAR Research and Test Department is greatly appreciated.
Additionally, the following persons and organizations contributed significant
amounts of in-kind support toward the completion of this document:
Beth Albertson Friedman & Bruya, Inc.
John Fitzgerald Massachusetts Department of
Environmental Protection
Elizabeth A. Harvey, Chevron Research and Technology
John Fetzer
G. Cornell Long United States Air Force,
Armstrong Laboratory
Ileana Rhodes Shell Development Company
George Sawyer Mobil Oil Corporation
Wade Weisman Air Force Research Laboratory,
Operational Toxicology Branch
Donna J. Vorhees, Menzie-Cura & Associates, Inc.
Chris M. Long
xi

1. PURPOSE
The chemical composition of petroleum products is complex and may change over
time following release into the environment. These factors make it difficult to
select the most appropriate analytical methods for evaluating environmental
samples. The Total Petroleum Hydrocarbon Criteria Working Group (the
“Working Group”) prepared this volume to assist site managers, risk assessors, reg-
ulators and others who may not have expertise in analytical chemistry in under-
standing the complexities of petroleum hydrocarbon characterization. This
volume describes petroleum analytical methods and what they can and cannot tell
you about environmental media (e.g., soil, groundwater, and surface water) at
petroleum contaminated sites. The information provided in this volume will help
the reader to accurately interpret results from petroleum analytical methods. This
volume is not meant to be a comprehensive text on analytical methods and does
not recommend or mandate any particular method. The goal of the Working
Group is to provide the technical information needed by regulators, risk assessors,
and site managers to implement health risk-based decisions at petroleum contam-
inated sites.
2. INTRODUCTION
There are a significant number of petroleum hydrocarbon impacted sites across
the United States resulting from a wide range of past industrial, military, and petro-
leum production, and distribution practices. Difficulties in evaluating and remedi-
ating these sites arise from the complexity of the regulatory, scientific, and eco-
nomic issues regarding impacted soil and water. Most investigations involving
petroleum hydrocarbons are regulated by the states with different requirements in
methodologies, action levels, and cleanup criteria. The chemical composition of
petroleum products is complex and varied and changes over time and distance
when released to the environment. These factors make it difficult to select the most
appropriate analytical test methods for evaluating environmental samples and to
accurately interpret and use the data.
The Working Group developed this volume to provide summary information
about analytical techniques and methods for total petroleum hydrocarbons (TPH),
petroleum by chemical class and boiling point ranges, and individual petroleum
constituents. Newer fraction-based analytical approaches also are discussed.
Discussion centers on analytical methods for soil, sediment, and water because
most published methods measure TPH in these media. However, much of the dis-
cussion in this volume is relevant to other environmental media, such as air.
Analytical methods for media such as sediment and air are often modified versions
of established methods for soil and water.
This volume is organized into the following principal sections:
Section 3: Overview of TPH
This section provides an overview of the historical measurement and use of
TPH data.
1

Section 4: Understanding The Petroleum Analytical Process: From Sample Collection
To Measurement
This section provides a general overview of the analytical process characteris-
tics that are common to most methods for quantifying TPH, individual petro-
leum constituents, and petroleum fractions.
Section 5: Total Petroleum Hydrocarbon (TPH) Measurement: Detailed Review Of
Selected Analytical Methods
This section describes analytical methods for TPH that are based on four main
analytical techniques: gas chromatography, infrared spectrometry, gravimetry,
and immunoassay measurements.
Section 6: Petroleum Group Type Measurement: Detailed Review Of Selected
Analytical Methods
This section describes analytical methods for petroleum groups that are based
on two main analytical techniques: thin layer chromatography and immunoas-
say measurements. Petroleum groups include different categories of hydro-
carbons such as saturates, aromatics, and polars/resins.
Section 7: Individual Petroleum Constituent Measurement: Detailed Review of Selected
Analytical Methods
This section describes analytical methods for individual petroleum con-
stituents that are based on four main analytical techniques: gas chromatogra-
phy with photoionization detection, gas chromatography with flame ioniza-
tion detection, high performance liquid chromatography, and gas chro-
matography with mass spectrometry detection. Individual petroleum con-
stituents often include benzene, carcinogenic polycyclic aromatic hydrocar-
bons, and other compounds commonly associated with petroleum.
Section 8: Evolving Methods For Petroleum Hydrocarbon Fractions
This section provides an introduction to several newer analytical methods for
quantifying petroleum fractions rather than TPH, groups or individual con-
stituents. It explains why such methods are being developed for evaluating
human health risk at petroleum contaminated sites.
3. OVERVIEW OF TPH
This section presents a historical perspective on the use of TPH in evaluating petro-
leum contaminated sites. Technical terms used in this section are explained in
greater detail in subsequent sections. The use of TPH concentrations to establish
target cleanup levels for soil or water is a common approach implemented by regu-
latory agencies in the United States. Approximately 75% of the states use TPH-based
cleanup criteria. Because these values have become such key remediation criteria, it
is essential that everyone using TPH data - environmental coordinators, field per-
sonnel, and regulators - be knowledgeable about the various analytical methods. It is
important to know that minor method deviations may be found from state to state.
TPH is sometimes referred to as mineral oil, hydrocarbon oil, extractable hydro-
carbon, and oil and grease. There are many analytical techniques available that
measure TPH concentrations in the environment. No single method measures the
2

entire range of petroleum-derived hydrocarbons .Because the techniques vary in
the way hydrocarbons are extracted, cleaned up, and detected, they each measure
slightly different subsets of the petroleum-derived hydrocarbons present in a
sample. See Section 4. The definition of TPH depends on the analytical method
used because the TPH measurement is the total concentration of the hydrocarbons
extracted and measured by a particular method. The same sample analyzed by dif-
ferent TPH methods may produce different TPH values. For this reason, it is
important to know exactly how each determination is made. Interpretation of the
results depends on understanding the capabilities and limitations of the selected
method. If used indiscriminately, TPH data can be misleading and could lead to an
inaccurate assessment of risk.
There are several reasons why TPH data do not provide ideal information for
investigating sites and establishing target cleanup criteria. For example, use of the
term TPH suggests that the analytical method measures the combined concentra-
tion of all petroleum-derived hydrocarbons, thereby giving an accurate indication
of site contamination. But this is not always the case. Furthermore, target cleanup
levels based on TPH concentrations implicitly assume (1) the TPH result is an
accurate measurement of petroleum-derived hydrocarbon concentration, and (2)
the TPH result indicates the level of risk associated with the contamination. These
assumptions are not correct due to many factors including the nonspecificity of
some of the methods used and, the complex nature of petroleum hydrocarbons
and their interaction with the environment over time.
One significant difficulty in measuring TPH for different product types is the
fact that the boiling ranges and carbon number ranges of refined petroleum prod-
ucts often overlap. Refined petroleum products are primarily manufactured
through distillation processes that separate fractions from crude oil by their boiling
ranges. See Appendix III — Refinery Flow Diagrams. Manufacturing processes may
also increase the yield of low molecular weight fractions, reduce the concentration
of undesirable sulfur and nitrogen components, and incorporate performance
enhancing additives. Additionally, because it is impossible to identify all con-
stituents of a petroleum product, these constituents are often described by their
boiling point ranges. Because distillations are not capable of producing sharp dis-
tinctions in boiling point cutoffs, there is overlap between distillate fractions. The
boiling point ranges correlate to carbon number and the higher the carbon
number, the higher the boiling point. However, structure will also influence
boiling point. Branched and aromatic compounds of the same carbon number
differ in boiling point from their corresponding n-alkane analogs. For these
reasons, boiling point actually defines an approximate carbon range.
Figure 1 shows the relationship between boiling range and carbon number for
some common petroleum products. This figure clearly shows the overlap between
carbon ranges of different products as well as the overlap in corresponding ana-
lytical methods. For example, Figure 1 shows that an analytical method designed
for gasoline range organics may report some of the hydrocarbons present in diesel
fuel. The same is also true for analytical tests for diesel range organics that will
identify some of the hydrocarbons present in gasoline contaminated soils. A more
detailed discussion of boiling point and carbon number classification as well as a
discussion of petroleum product composition, specification, product additives, and
weathering is provided in Appendix II: Characterization of Petroleum Products.
3

4
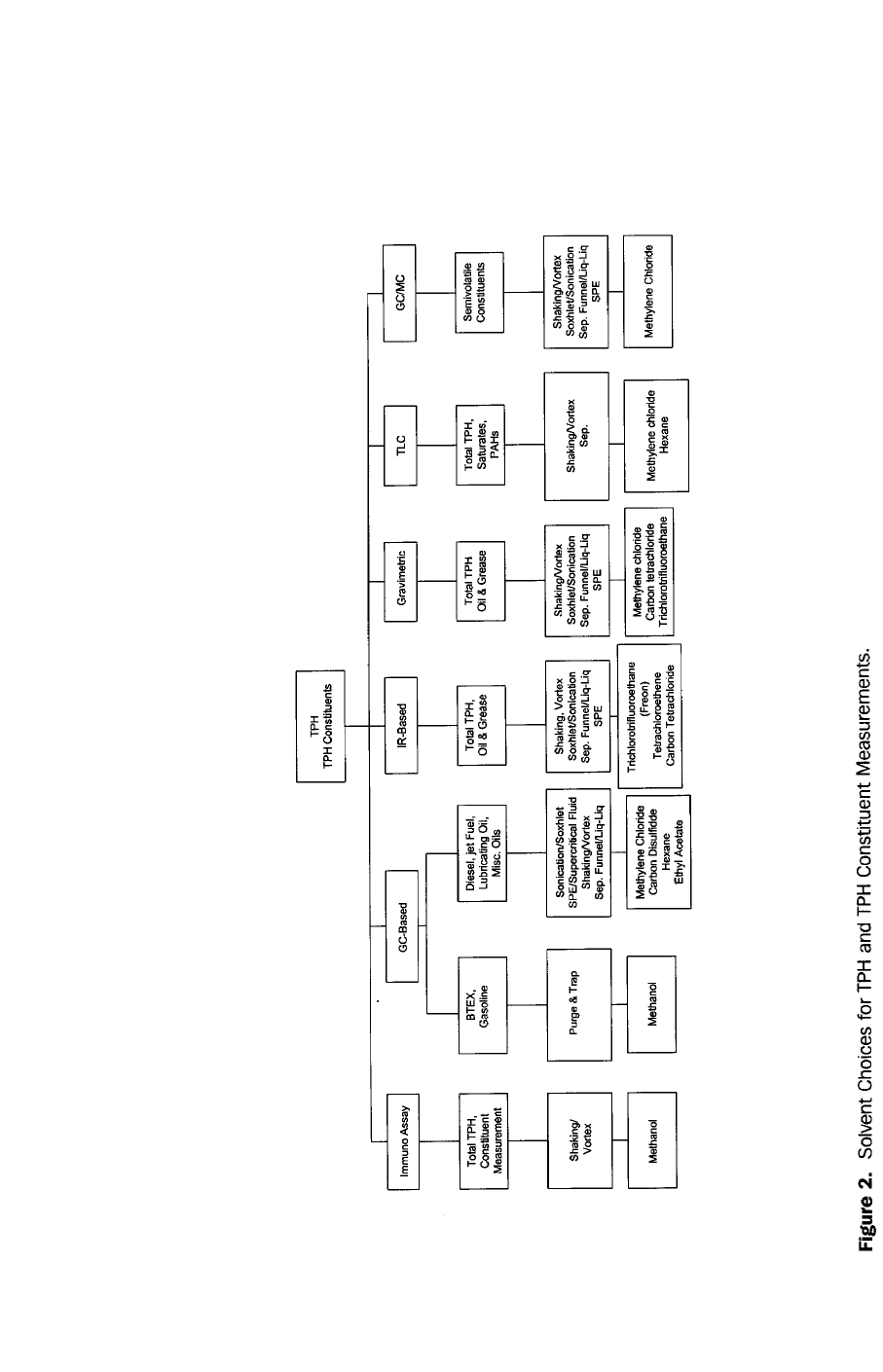
Figure 1. Summary of Petroleum Product Types and TPH and TPH Analytical Methods with Respect to Approximate Carbon Number
and Boiling Point Ranges.

Ambiguous terminology associated with TPH methods presents additional diffi-
culty in interpreting results. Each method has its own designation. For example,
TRPH stands for total recoverable petroleum hydrocarbons, DRO stands for diesel
range organics, GRO stands for gasoline range organics, and TPH-G stands for
total petroleum hydrocarbons-gasoline. Frequently a method name that cites a
product - “gasoline” or “diesel” - only implies a carbon range. For example, TPH-G
does not necessarily imply that gasoline is present. These abbreviations may imply
different carbon ranges to different laboratories or states. TPH methods are not
optimized to identify product type. Even with improved, more detailed analytical
methods, identification of aged products may prove difficult.
There is a reason for the availability of a large number of TPH measurement
techniques. Because petroleum and petroleum-derived products are such complex
mixtures, there is no single “best” method for measuring all types of petroleum
contamination. As shown in Figure 1, some methods are appropriate for gasoline-
contaminated samples while others are designed for heavier hydrocarbon contam-
ination such as jet or diesel fuel. Some methods measure more compounds than
other methods because they employ more rigorous extraction techniques or more
efficient solvents. Other methods are subject to interferences from naturally occur-
ring materials such as animal and vegetable oils, peat moss, dried grass, or humic
material in topsoil, which may result in artificially high reported TPH concentra-
tions. Some methods use cleanup steps to minimize the effect of nonpetroleum
hydrocarbons, with variable success. Ultimately, methods are limited by the extrac-
tion and cleanup efficiency and measurement device detection limits.
The choice of a specific method should be based on compatibility with the par-
ticular type of hydrocarbon contamination to be measured. The choice may
depend upon state regulatory requirements for the type of hydrocarbon contami-
nation suspected to be present.
3.1 RISK IMPLICATIONS
TPH concentration data cannot be used to quantitatively estimate human health
risk. The same concentration of TPH may represent very different compositions
and very different risks to human health and the environment. For example, two
sites may have TPH measurements of 500 ppm but constituents at one site may
include carcinogenic compounds while these compounds may be absent at the
other site. The risk at a specific site will change with time as contaminants evapo-
rate, dissolve, biodegrade, and become sequestered. A valid correlation between
TPH and risk would have to be site- and time-specific, related to a single spill, and,
even then, the correlation might not be the same around the periphery of a plume
where the rate of compositional change accelerates.
Although the utility of TPH data for risk assessment is minimal, it is an inexpen-
sive tool that can be used for three purposes: (1) determining if there is a problem;
(2) assessing the severity of contamination; and (3) following the progress of a
remediation effort. If TPH data indicate that there may be significant contamina-
tion of environmental media, other data can be collected so that harm to human
health can be quantitatively assessed. These other data can include target analyte
concentration data and petroleum fraction concentration data obtained using new
fraction-based analytical methods developed by the Working Group and others.
5

4. UNDERSTANDING THE PETROLEUM ANALYTICAL PROCESS: FROM
SAMPLE COLLECTION TO MEASUREMENT
This volume focuses on three types of petroleum analytical methods:
• methods that measure a TPH concentration;
• methods that measure a petroleum group type concentration; and
• methods that measure individual petroleum constituent concentrations.
These three types of methods measure different petroleum hydrocarbons that
might be present in petroleum-contaminated environmental media. TPH methods
generate a single number that represents the combined concentration of all petro-
leum hydrocarbons in a sample, which are measurable by the particular method
(See discussion in Section 3 regarding limitations of TPH data). Petroleum group
type methods separate and quantify different categories of hydrocarbons (e.g., sat-
urates, aromatics, and polars/resins). The results of petroleum group type analyses
can be useful for product identification because different products (e.g., gasoline,
fuel oil no.2, and jet fuel) can have characteristic levels of various petroleum
groups (see Appendix II for a detailed characterization of petroleum products).
Individual constituent methods quantify concentrations of specific compounds
that might be present in petroleum-contaminated samples, such as benzene, ethyl-
benzene, toluene, and xylenes (BTEX), and polycyclic aromatic hydrocarbons
(PAHs). Concentration data for individual petroleum constituents can be used to
evaluate human health risk, provided the necessary toxicity data are available.
Although these three method types measure different petroleum hydrocarbon
categories, there are several basic steps that are common to the analytical process-
es for all methods, no matter the method type or the environmental matrix. This
section will focus on these basic steps. Sections 5, 6 and 7 review analytical methods
that can provide the three different types of petroleum concentration data.
Most of the common analytical steps are related to the separation of analytes of
interest from a sample matrix prior to their measurement. In general, these steps are:
• Collection and preservation - requirements specific to environmental matrix
and analytes of interest
• Extraction - separates the analytes of interest from the sample matrix
• Concentration - enhances the ability to detect analytes of interest
• Cleanup - may be necessary to remove interfering compounds
• Measurement - quantifies the analytes.
Each step affects the final result, and a basic understanding of the steps is vital
to data interpretation.
6

4.1 COLLECTION AND PRESERVATION OF ENVIRONMENTAL SAMPLES
The ability to collect and preserve a sample that is representative of the site is a
critically important step. Obtaining representative environmental samples is
always a challenge due to the heterogeneity of different sample matrices.
Additional difficulties are encountered with petroleum hydrocarbons due to the
wide range in volatility, solubility, biodegradation, and adsorption potential of
individual constituents.
Most site investigations for assessment of petroleum hydrocarbon contamination
in the environment are regulated by the states. However, sample collection and
preservation recommendations follow U.S. EPA guidelines. A summary of the most
commonly used guidelines is included in Table 1. It should be noted that there
might be additional requirements in any given state. Before a sample is collected,
the particular state requirements must be investigated. Because of holding time
considerations, the laboratory must be selected and notified prior to the collection
of the samples.
4.2 SAMPLE EXTRACTION
For most analyses, it is necessary to separate the analytes of interest from the matrix
(i.e. soil, sediment, and water). Extraction of analytes can be performed using one
or more of the following methods:
• Extracting the analytes into a solvent
• Heating the sample (used in the analyses of volatile compounds)
• Purging the sample with an inert gas (used in the analyses of volatile compounds).
There are a variety of common sample extraction techniques. See Table 2.
Soxhlet, sonication, supercritical fluid, subcritical or accelerated solvent, and
purge and trap extraction have been promulgated by the U.S. EPA as soil extrac-
tion methods. Headspace is recommended as a screening method. Shaking/vor-
texing is presently not approved by EPA, but is quite adequate for the extraction of
petroleum hydrocarbons in most environmental samples.
For these extraction methods, the ability to extract petroleum hydrocarbons
from soil and water samples depends on the solvent and the sample matrix.
Surrogates (compounds of known identity and quantity) are frequently added to
monitor extraction efficiency. Environmental laboratories also generally perform
matrix spikes (addition of target analytes) to determine if analytes are retained by
the soil or water matrix.
Solvents have different extraction efficiencies. Extracting the same sample in the
same manner by two different solvents may result in different concentrations. The
choice of solvents is determined by many factors such as cost, spectral qualities,
method regulations, extraction efficiency, toxicity, and availability. Methylene chlo-
ride has been the solvent of choice for many semivolatile analyses due to its high
extraction efficiency, low cost, and specification by many state regulatory methods.
Chlorofluorocarbon solvents such as trichlorotrifluoroethane (Freon 113) have
been used in the past for oil and grease analyses because of their spectral qualities
(they do not absorb in the 2930 cm-1 infrared measurement wavelength) and low
7

8
TABLE 1. U.S. EPA-Recommended Sampling Protocols
Sample Container
a
Analytical Analytical Holding
Parameter Method(s) Media Volume Type Preservatives
c
Time
Trph
b
EPA 418.1 (IR); water 1 liter Glass jar with acid fix pH<2; extract in 7 days;
Gravimetric; Teflon lined cap cool to 4°C analyze in 40 days
GC/FID soil 125 mL Wide mouth glass cool to 4°C extract in 7 days;
Teflon lined cap analyze in 40 days
Volatile Petroleum various water 40 mL Glass vial with acid fix pH<2; 14 days
Hydrocarbons (VPH)
d
Teflon lined septum cool to 4°C
soil 40 mL Glass vial with cool to 4°C
e
14 days
Teflon lined septum
Extractable Petroleum various water 1 liter Glass jar with acid fix pH<2; extract in 7 days;
Hydrocarbons (EPH)
f
Teflon lined cap cool to 4°C analyze in 40 days
soil 60 mL Wide mouth glass cool to 4°C extract in 7 days;
Teflon lined cap analyze in 40 days
BTEX EPA 8240/8260
h
water 40 mL Glass vial with acid fix pH<2; 14 days
EPA 8020/8021
h
Teflon lined septum cool to 4°C
EPA 624
g
, EPA 602
g
soil 40 mL Glass vial with cool to 4°C
e
14 days
EPA 524
g
Teflon lined septum
PAHs EPA 8270
h
water 1 liter Glass jar with acid fix pH<2; extract in 7 days;
EPA 8310
h
Teflon lined cap cool to 4°C analyze in 40 days
EPA 8100
h
soil 60 mL Wide mouth glass cool to 4°C extract in 7 days;
Teflon lined cap analyze in 40 days

9
TABLE 1. (Continued)
a
Minimum sampling volume may vary depending on specific method.
b
Refers to extractable hydrocarbons only.
c
ACID FIXATION: use 1:1 HCL to adjust pH of aqueous samples to less than 2. Add approx. 500µl (2-4 drops) to 40 mL aqueous sample vials; 5
mL to 1 liter aqueous sample jars. Add acid to vials before collecting sample; use gloves and eye protection when sampling. Other preserva-
tives, such as sulfuric acid or sodium bisulfate may also be used for this purpose.
d
Generally C
5
through C
10±2
hydrocarbons detectable through purge and trap or headspace analyses; includes most “Gasoline Range Organics”
(GRO) methodologies.
e
Some states/methodologies require field preservation of soil samples in methanol. In such cases, methanol must be purge and trap grade; typi-
cally, add 20 mL methanol to vials prior to sample collection. Use gloves and eye protection when sampling. Shipping of methanol subject to
DOT regulation.
f
Generally C
9
through C
28±7
hydrocarbons detectable through a solvent extraction process; includes most “Diesel Range Organics” (DRO) method-
ologies.
g
40 CFR Part 136.
h
SW-846 methodology.

10
TABLE 2. Comparison of Common Extraction Techniques
Extraction US SW-846 Extraction Compounds
Method Method Number Matrix Extracted Purpose
Separatory funnel 3510 water semivolatile laboratory
nonvolatile
Continuous liquid-liquid 3520 water semivolatile laboratory
nonvolatile
Solid phase extraction 3535 water semivolatile laboratory/
nonvolatile screening
Purge and trap 5030, 5035 water, soil volatile laboratory/
field preservation
Headspace 3810, 5021 water, soil volatile screening/
laboratory
Shake and vortex
a
soil volatile screening/
semivolatile laboratory
nonvolatile
Soxhlet 3540, 3541 soil semivolatile laboratory
nonvolatile
Sonication 3550 soil semivolatile laboratory
nonvolatile
Supercritical fluid 3560, 3561 soil semivolatile laboratory
nonvolatile
Subcritical fluid 3545 soil semivolatile laboratory
nonvolatile
a
Not an EPA SW-846 approved method.
11

human toxicity. The EPA is phasing out use of chlorofluorocarbons, however, due
to their detrimental effects on stratospheric ozone. Tetrachloroethene and carbon
tetrachloride are possible replacements. Methanol is the most common solvent
used to preserve and extract volatiles such as BTEX in soils. Figure 2 illustrates the
typical solvents used for different analyses.
Water Samples
Water extraction methods in common use include the following:
For Volatiles:
• Purge and trap
• Headspace
For Semivolatiles:
• Separatory funnel extraction
• Continuous liquid-liquid extraction
• Solid phase extraction.
Volatile compounds (gasoline, solvents) in water are generally separated from
their matrix by purging with an inert gas and trapping the compounds on a
sorbent (EPA 5030, purge and trap analysis). The sorbent is later heated to
release the volatile compounds, and a carrier gas sweeps the compounds into a
gas chromatograph.
Headspace analysis is recommended as a screening method by EPA (Methods
3810, and 5021), although it performs well in particular situations, especially field
analysis. In this method, the water sample is placed in a closed vessel with a head-
space and heated to drive volatiles into the gas phase. Addition of salts or acids may
enhance this process. In headspace analysis, instrument contamination is mini-
mized because only volatile compounds are introduced into the instrument.
Samples containing heavy oils and high analyte concentrations can severely conta-
minate purge and trap instrumentation.
The most commonly used water extraction method for semivolatiles is EPA
Method 3510, separatory funnel extraction. The sample is poured into a funnel-
shaped piece of glassware, solvent is added, and the mixture is shaken vigorously.
After layer separation, the extract (i.e., the solvent layer) is removed, filtered, dried
with a desiccant, and concentrated. Multiple extractions on the same sample may
increase overall recovery.
Another commonly used water extraction method for semivolatiles is EPA 3520,
continuous liquid-liquid extraction. Rather than shaking solvent with the water
sample, the solvent is continuously heated, nebulized (broken into small droplets),
and sprayed on top of the water. Liquid-liquid extraction is excellent for samples
containing emulsion-forming solids, but it is more time-consuming than separato-
ry funnel extractions.
Solid phase extraction (SPE, EPA Method 3535) also can be used for extraction
and concentration of semivolatile material. The technique involves passing the
water sample through a cartridge or disk containing an adsorbent such as silica or
12

alumina. The adsorbent is often coated with compounds that impart selectivity for
particular products or analytes such as PAHs. After extraction, the analytes are sep-
arated from the solid phase by elution with a small amount of organic solvent. A
variant of SPE involves dipping a sorbent-coated fiber into the water (solid phase
micro-extraction, or SPME). Adsorbed analytes are thermally desorbed directly
into a heated chromatographic injection port. SPE uses much less solvent and
glassware than separatory funnel and liquid-liquid extraction.
Soil Samples
Soil extraction methods in common use include the following:
For Volatiles:
• Purge and trap
• Headspace
For Semivolatiles:
• Shaking or vortexing (can also be used for volatiles)
• Soxhlet
• Sonication
• Supercritical fluid
• Subcritical fluid.
Volatile compounds (e.g., BTEX and gasoline) may be solvent-extracted from
soil. EPA Method 5035, purge and trap analysis, specifies a methanol extraction,
which is usually done by mechanical shaking of the soil with methanol. A portion
of the methanol extract is added to a purge vessel and diluted in reagent grade
water. The extract is then purged similar to a water sample.
Headspace analysis, (EPA Methods 3810 and 5021), also works well for analyzing
volatiles in soils. The soil is placed in a headspace vial and heated. Salts can be
added to more efficiently drive out the volatiles from the sample into the head-
space of the sample container. Similar to water headspace analysis, the soil head-
space technique is useful when heavy oils and high analyte concentrations are
present which can severely contaminate purge and trap instrumentation.
Detection limits are generally higher for headspace analysis than for purge and
trap analysis.
The simplest method to separate semivolatile compounds from soils is to shake
or vortex (vigorous mechanical stirring) soil with a solvent. Adding a desiccant to
the soil/solvent mixture can help to break up soil and increase the surface area.
The extract can be analyzed directly. Simple shaking is quick and easy, making it an
excellent field extraction technique. However, extraction efficiency may vary
depending on soil type.
Soxhlet extraction (EPA Method SW-846 3540) is a very efficient extraction
process which is commonly used for semivolatiles. Solvent is heated and refluxed
(recirculated) through the soil sample continuously for 16-24 hours or overnight.
This method generates a relatively large volume of extract that needs to be con-
centrated. Thus, it is more appropriate for semivolatiles than for volatiles.
13

Sonication extraction (EPA Method SW-846 3550) can also be used for semi-
volatiles, it involves the use of sound waves to enhance analyte transfer from sample
to solvent. Sonication is a faster technique than Soxhlet extraction, and it also can
require less solvent.
Supercritical fluid extraction (SFE), (EPA Methods 3540 for Total Recoverable
Petroleum Hydrocarbons and 3561 for PAHs), is applicable to the extraction of
semivolatiles. SFE involves heating and pressuring a mobile phase to supercritical
parameters (having properties of gas and liquid). The supercritical fluid is passed
through the soil sample, and the analytes are concentrated on a sorbent or trapped
cryogenically. The analytes are eluted with a solvent and analyzed with conven-
tional techniques. CO
2
is the most popular mobile phase.
Proposed EPA Method 3545, Accelerated Solvent Extraction, has extraction effi-
ciencies comparable to Soxhlet extractions. This method is sometimes referred to
as subcritical fluid extraction. Conventional solvents such as methylene chloride are
heated and pressurized, then passed through the soil sample (this technique also
works for sludges and sediments). Subcritical fluid extraction has the advantage of
requiring smaller solvent volumes than traditional solvent extraction techniques.
Free Phase Hydrocarbon Samples
In some releases of petroleum products to the environment where free phase
product is found, hydrocarbon material can be collected directly for characteriza-
tion. The ability to analyze free product greatly aids the determination of product
type and potential source. The samples may be diluted prior to analysis. EPA Method
SW-846 3580, waste dilution, gives some guidelines for proper dilution techniques.
4.3 CONCENTRATION OF SAMPLE EXTRACT
Extracts are generally filtered, dried with desiccant, and concentrated before analy-
sis. Concentration of the extract may allow for lower sample detection limits.
Frequently, sample extracts must be concentrated to obtain detection limits low
enough to meet regulatory action limits. Concentration may be achieved by:
For Volatiles:
• Sorbent trapping
• Cryogenic trapping
For Semivolatiles:
• Snyder column
• Kuderna Danish concentrator
• Nitrogen evaporation
• Vacuum.
The trapping step in a purge and trap analysis is essentially a concentration step.
Analytes are purged from the matrix into a gas stream and captured on a sorbent
trap. The analytes are released by heating the trap. Some laboratories use cryo-
genic trapping in place of sorbent trapping. A very cold material such as liquid
14

nitrogen surrounds a sample loop. As analytes are purged and swept through the
sample loop, they freeze in the sample loop. The analytes are released when the
trap is heated.
Snyder columns are designed to allow highly volatile solvents to escape while
retaining semivolatile analytes of interest. Snyder columns are generally fitted onto
the tops of flasks containing extracts. The column design permits solvent to escape
as the flask is heated. The analytes of interest condense from a gas to a liquid phase
and fall back down into the solvent reservoir. The Kuderna-Danish concentrator is
a Snyder column with a removable collection tube attached to the bottom. As
solvent is evaporated, the extract is collected in the collection tube.
As an alternative to a Snyder column, the sample extracts may be concentrated with
nitrogen evaporation by directing a slow stream of the gas over the extract surface at
room temperature, resulting in minor loss of volatiles. Placing the extract container
in warm water helps to speed the process, but then some loss of volatiles can occur.
Concentration by evaporating excess solvent with a vacuum is not very common
in environmental laboratories. Many semivolatile analytes are lost in the proce-
dure. Additionally, evaporating as a means of concentrating the sample cannot be
used if the goal is to detect volatile analytes.
4.4 CLEANUP OF SAMPLE EXTRACT
Cleanup steps can be an important component of infrared (IR)-based and gravi-
metric methods because these methods are very sensitive to nonpetroleum
hydrocarbon interferences. Cleanup steps are less commonly utilized for gas
chromatography (GC)-based methods because experienced GC analysts can rec-
ognize the presence of interfering compounds (e.g., animal- and vegetable-
derived hydrocarbons).
Cleanup steps are not always a part of the petroleum analytical process, but when
they are necessary, the goals of extract cleanup steps typically include one or more
of the following:
• Removal of nonpetroleum compounds
• Isolation of a particular petroleum fraction
• Concentration of analytes of interest.
The techniques employed to extract the analytes of interest can frequently
extract interfering compounds. Polar compounds such as animal and plant fats,
proteins, and small biological molecules may be improperly identified as petrole-
um constituents. Extract cleanup techniques can be used to remove them. In an
ideal situation, only interfering compounds are removed. In reality, some polar
petroleum constituents can also be removed.
Two techniques are used to clean petroleum extracts. In one technique, inter-
fering compounds are removed by passing the extract through a glass column
filled with sorbent. A second technique is to swirl the extract with loose sorbent,
then remove the sorbent by filtration.
Three of the EPA-promulgated cleanup techniques involve trapping the interfer-
ing compounds on a sorbent column. EPA SW-846 3611 is an alumina cleanup
designed to remove interfering compounds and to fractionate petroleum wastes
15

into aliphatic, aromatic and polar fractions. The fractions can be analyzed sepa-
rately or combined for a total TPH measurement. EPA SW-846 3630, silica gel
cleanup, is the most common cleanup technique used on extracts designated for
PAHs and phenol analyses. Variations of this technique are used to clean EPA
Method 418.1 extracts before IR analysis. EPA Method SW-846 3640, gel-permeation
cleanup, works on the principle of size exclusion. Large macromolecules such as
lipids, polymers, and proteins are removed from the sample extract. Extracts
obtained from soils with high biological activity may be cleaned by this method.
There are two noncolumn EPA-promulgated cleanup techniques. EPA SW-846
3650, acid-base partition can be used to separate the base/neutral and acid com-
ponents by adjusting pH. Very often, only one fraction contains the compounds of
interest. This technique is often used before alumina column cleanup to remove
the acid components. EPA Method SW-846 3660 is used for sulfur removal. This
technique uses copper, mercury, and tetrabutylammonium sulfite as desulfuriza-
tion compounds. Sulfur is a common interfering compound for petroleum hydro-
carbon analysis, particularly for sediments. Sulfur-containing compounds are very
common in heavy fuels and crudes and on refinery sites. Elemental sulfur is often
present in anaerobically biodegraded fuels. High levels of sulfur may be measured
as “TPH” by some techniques if the cleanup technique is not used.
Solid phase cartridges for sample cleanup are available from chromatography
suppliers. These cartridges are available in a wide variety of adsorbents with unique
chemical selectivities; however, they have limited capacity.
There can be several limitations to various cleanup steps. Reasons for decreased
effectiveness of cleanup procedures include:
• Sample loading may exceed the capacity of cleanup columns and cartridges.
• Nonpetroleum compounds may have chemical structures similar to petrole-
um compounds and may behave like a petroleum compound. These com-
pounds evade cleanup.
• The cleanup may not have been done properly (poor technique).
• Some analytes of interest may be removed.
• There may be cases in which no single cleanup technique removes all
interferences.
For example, Table 3 shows the results of serial silica gel cleanups of EPA Method
418.1 extracts of plant materials. These results illustrate the limited effectiveness of
multiple silica gel cleanups for the removal of biogenic hydrocarbon interferences.
4.5 MEASUREMENT
Once the sample preparation is complete, there are several approaches for detect-
ing and quantifying petroleum hydrocarbons:
• Total petroleum hydrocarbon (TPH) measurement
• Petroleum group type measurement
• Individual component measurement.
16

17
TABLE 3. Effect of Silica Gel on Removing Biogenic Hydrocarbon Interferences
from Vegetative Materials (Freon-113 Extracts)
a
TPH concentrations given in ppm (mg/kg)
Prior to Addition After First Addition After Second Addition After Third Addition
Vegetable Material of Silica Gel of Silica Gel of Silica Gel of Silica Gel
Fresh pine needles 16,000 1,700 1,400 —
Pine bark 2,400 380 370 —
Pine needle compost 1,200 70 67 —
Maple tree seeds 7,100 1,600 1,500 —
Oak leaves dried 18,000 4,800 4,600 —
Grass, dried 14,000 4,500 2,700 2,600
Gall nuts 9,700 4,500 1,300 1,200
a
Source: Data from “EPA Method 418-1 Total Recoverable Petroleum Hydrocarbons by IR”, Groundwater Analytical Bulletin,
(Buzzards Bay: Groundwater Analytical Inc. 1991).

Total Petroleum Hydrocarbon (TPH) Measurement
Total petroleum hydrocarbon (TPH) measurements are conducted to determine
the total amount of hydrocarbon present in the environment. There are a wide
variety of TPH methods. In practice, TPH is defined by the method used to
analyze it. Different methods often give different results because they are
designed to extract and measure slightly different subsets of petroleum hydrocar-
bons. No single method gives a precise and accurate measurement of TPH for all
types of contamination. The four most commonly used TPH testing methods
include gas chromatography (GC), infrared spectrometry (IR), gravimetric analy-
sis, and immunoassay.
Petroleum Group Type Measurement
Petroleum group type measurements are conducted to determine amounts of
various petroleum compound classes (e.g., saturates, aromatics, and polars/resins)
present in a petroleum contaminated samples. Compound classes are discussed in
detail in Appendix I. This type of measurement is sometimes used to identify fuel
type or to track plumes. It may be particularly useful for heavier hydrocarbons such
as tar and asphalt. Group type test methods include multidimensional gas chro-
matography (not often used for environmental samples), high performance liquid
chromatography (HPLC), and thin layer chromatography (TLC).
Petroleum Constituent Measurement
Methods that analyze individual compounds (e.g., BTEX and PAHs) are generally
run to detect the presence of an additive or to provide concentration data needed
to estimate human health risk associated with individual compounds. Common
constituent measurement techniques include gas chromatography with second
column confirmation, gas chromatography with multiple selective detectors and
gas chromatography with mass spectrometry detection (GC/MS).
5. TOTAL PETROLEUM HYDROCARBON (TPH) MEASUREMENT:
DETAILED REVIEW OF SELECTED ANALYTICAL METHODS
A TPH method generates a single number quantifying the amount of petroleum
that is measured by the specified technique. The most popular TPH methods are
based on gas chromatographic (GC), infrared (IR), or gravimetric analytical tech-
niques. GC-based methods are currently the preferred laboratory methods for
TPH measurement because they detect a broad range of hydrocarbons, they
provide both sensitivity and selectivity, and they can be used for TPH identification
as well as quantification. IR-based methods have been widely used in the past for
TPH measurement because they are simple, quick and inexpensive. However, their
use is currently decreasing due to the worldwide ban on Freon production
(needed for sample extraction and measurement), the nonspecificity of these
methods, and their inability to provide any information on TPH identification and
potential risk. Gravimetric-based methods are also simple, quick, and inexpensive,
18

but they suffer from the same limitations as IR-based methods. Gravimetric-based
methods may be useful for very oily sludges and wastewaters, which will present
analytical difficulties for other more sensitive methods. Immunoassay TPH
methods are gaining popularity for field testing because they offer a simple, quick
technique for in-situ TPH quantification.
The following four sections provide detailed descriptions of these TPH methods.
Each section provides an overview of the analytical technique, example methods,
the purpose of the method (i.e., what it is intended to measure), and key interfer-
ences/limitations. Table 4 briefly summarizes this information for each TPH ana-
lytical method for quick reference. Appendix IV includes tables that provide more
detailed information about published GC-based and non-GC TPH methods. These
tables provide method-specific information for EPA and state methods, including
recommendations for method use, common interferences, procedural notes,
advantages, and disadvantages.
5.1 GAS CHROMATOGRAPHY (GC) TPH METHODS
For GC-based methods, TPH is defined as anything extractable by a solvent or
purge gas and detectable by gas chromatography/flame ionization detection
(GC/FID) within a specified carbon range. The primary advantage of GC-based
methods is that they provide information about the type of petroleum in the
sample in addition to measuring the amount. Identification of product type(s) is
not always straightforward, however, and requires an experienced analyst of petro-
leum products. Detection limits are method- and matrix-dependent and can be as
low as 0.5 mg/L in water or 10 mg/kg in soil.
Overview of the Technique
Gas chromatography is a technique that separates mixtures. “A mixture of chemi-
cals is separated into its individual components as the sample travels through a
column in the gas chromatograph. Separation is achieved by a combination of
factors including boiling point, polarity, and affinity differences among the differ-
ent components in the sample. The time a compound spends on a specific column
is called the retention time and it is reproducible. The retention time is character-
istic of a compound under given experimental parameters and specified column.
As the separated components elute from the column, they are detected” (Swallow
et al., 1988). The detector signal is proportional to the amount of compound
present.
Chromatographic columns are commonly used to determine TPH compounds
approximately in the order of their boiling points. Compounds are detected with
a flame ionization detector, which responds to virtually all compounds that can
burn. The sum of all responses within a specified range is equated to a hydrocar-
bon concentration by reference to standards of known concentration.
Two techniques are most commonly used to get the samples into the column.
• Purge and trap systems purge components out of water or water/methanol by
bubbling gas through the liquid. The components are concentrated on a very
short intermediate column or “trap,” which is heated to drive them onto the
19

20
TABLE 4. General Summary of Analytical Methods for TPH Measurement
Typical Typical
Method Type/ Products Carbon Published
Method Name Environmental Media Detected Ranges Detected Methods
GC-Based TPH methods primarily laboratory but also primarily gasolines, diesel fuel, normally between C6 and EPA Method 8015B,
field applications- can be and fuel oil #2 - can be C25 or C36 (can be modified state-modified 8015
adapted for all media modified for heavier hydro- for higher carbon numbers) methods
carbon mixtures (e.g., lubricating
oils, heavy fuel oils)
IR-Based TPH methods laboratory and field screening primarily diesel and fuel oils most hydrocarbons with EPA Method 418.1
- most appropriate for water exception of volatile and
and soil very high hydrocarbons
Gravimetric TPH methods laboratory - most appropriate most appropriate for heavier anything that is extractable EPA Method 9071;
for wastewaters, sludges, petroleum products (e.g., crude (with exception of volatiles EPA proposed Method
and sediment oils, lubricating oils, etc.) which are lost) 1664
Immunoassay TPH field screening - most various products (but yields aromatic hydrocarbons EPA Method 4030
methods appropriate for soil and water only screening numbers) (e.g., BTEX, PAHs)

21
TABLE 4.
Detector Approximate Key Interferences/
Type Detection Limits Advantages Limitations
GC/FID can be as low as 0.5 mg/L can detect broad range of normally cannot detect
in water, 10 mg/kg in soil hydrocarbon compounds; simple compounds below C6;
and sensitive; can provide infor- may not detect polar
mation (e.g., a chromatogram) for hydrocarbons (e.g.,
product identification alcohols, ethers, etc.);
chlorinated solvents
may be quantified as TPH
IR spectrometer 1 mg/L in water; technique is simple, quick, Freon is now banned;
10 mg/kg in soil and inexpensive lack of specificity; low
sensitivity; high loss of
volatiles; poor extraction
of high molecular weight
hydrocarbons; prone to
interferences; provides
quantitation only
Gravimetric balance 5 to 10 mg/L in water; technique is simple, quick, Freon is banned,
50 mg/kg in soil and inexpensive although other solvents
are available; lack of
specificity; low sensitivity;
high loss of volatiles;
prone to interferences;
provides quantitation only
Portable test kit 200 to 500 ug/L in water; technique is simple, quick, low sensitivity; can
10 to 500 mg/kg in soil inexpensive, and can be done detect interferences; pri-
in the field marily only measure
aromatics; low accuracy
and precision; should
only be used as screening
measurement; provides
quantitation only

analytical column where they are separated. Hydrocarbons from C
5
through
about C
12
can be analyzed using this technique. Purge and trap sample intro-
duction is used for light products such as gasoline and condensate.
• Direct injection involves taking the hydrocarbon, diluted hydrocarbon, or
an extract of hydrocarbon into a syringe and injecting it into the gas chro-
matograph. This technique can be used for any type of hydrocarbon, but it
is most frequently used for distillates, lube oils and crude oils.
Headspace sample introduction can be used for the determination of light
hydrocarbons, and it is most often used for field screening.
Example Methods
Prior to 1997, EPA SW-846 Methods 8015 and 8015A were often quoted as the
source of GC-based TPH methods, commonly referred to as “Modified 8015.”
However, the original 8015 methods were titled “Nonhalogenated Volatile
Organics” and were designed to measure a short target list of chemical solvents
rather than petroleum hydrocarbons. Because there was no universal method for
petroleum hydrocarbons, each state specified its own version. The recent Update
III of EPA SW-846, 3rd Ed. includes a new Method 8015B titled “Nonhalogenated
Volatile Organics Using GC/FID”, with guidance for the analysis of gasoline and
diesel range organics. Whether the new method will replace the many techniques
currently in use is uncertain. The current individual methods differ in procedure,
compounds detected, extraction techniques and extraction solvents used. Some
methods may include a cleanup step to remove biogenic (bacterial or vegetation-
derived) material while others do not. The methods have in common a boiling
point-type column and a flame ionization detector.
Some regulatory agencies specify two GC-based TPH methods. Selection of a
method depends on the type of hydrocarbon suspected to be in the sample:
• If gasoline is suspected to be the sole contaminant, the TPH method will use
purge/trap sample introduction. Many of these methods are referred to as volatile
range organics - TPHV, TPH-G, or GRO - gasoline range organics. Typically, gaso-
line or a synthetic mixture will be used to prepare calibration standards.
• If heavier petroleum fractions (diesel, middle distillates, motor oil) are the
contaminants, the analysis will use direct injection and hotter oven tempera-
tures. Many of these methods are referred to as extractable range organics,
TPH-D or DRO - diesel range organics. Typically, diesel fuel #2 or a synthet-
ic mixture will be used to prepare calibration standards. Jet fuel or lube oil
may be used when appropriate.
• Mixtures or unknown contamination may require both volatile range and
extractable range analyses. Alternately, a single injection can be used to
analyze the whole sample, but the extraction method must not use a solvent
evaporation step. Reporting limits for a single injection method are approx-
imately 20 mg/L.
22

What Do GC Methods Measure?
A GC/FID will detect any hydrocarbons that elute from the column and burn. The
analog signal from the detector is called a chromatogram. GC/FID methods specify
a certain portion of the chromatogram (a “window” or carbon number range) for
quantification. The carbon number range will approximate that of the fuel of inter-
est - gasoline, diesel, or heavier hydrocarbon. The carbon number range specified
for each fuel may differ from state to state. Volatile compounds that elute before the
solvent peak (usually those < C
6
) are typically not measured.
GC-based methods can be broadly used for different kinds of petroleum releas-
es but are most appropriate for detecting nonpolar hydrocarbons with carbon
numbers between C
6
and C
25
or C
36
.
Many lube oils contain molecules with more than 40 carbon atoms. Crude oils
may contain molecules with 100 carbons or more. These heavy hydrocarbons are
outside the detection range of the more common GC-based TPH methods, but spe-
cialized gas chromatographs are capable of analyzing such heavy molecules.
Accurate quantification depends on adjusting the chromatograph to reach as high
a carbon number as possible, then running a calibration standard with the same
carbon range as the sample. The lab must also check for mass discrimination, a ten-
dency for heavy molecular weight hydrocarbons to be retained in the injection
port. Labs should be notified if a sample is suspected to be heavy oil, or to contain
a mixture of light and heavy oils, so that they can use the appropriate GC method.
Gravimetric or IR methods are often preferred for very heavy samples. They can
even be used as a check on GC/FID results if it is suspected that heavy molecular
weight hydrocarbons are present but are not being detected. Laboratories should
flag data if heavy material is observed in the chromatogram, even if this material
cannot be quantified.
Calibration standards vary. Most methods specify a gasoline calibration standard
for volatile range TPH and a diesel fuel #2 standard for extractable range TPH.
Some methods use synthetic mixtures for calibration. Because most methods are
written for gasoline or diesel fuel, TPH methods may have to be adjusted to
measure contamination by heavier hydrocarbons - lubricating oils, heavy fuel oils,
or crude. Such adjustments may entail use of a more aggressive solvent, a wider GC
“window” - up to C
36
or more - and a different calibration standard that more
closely resembles the “heavier” contamination.
GC-based methods can be modified and fine-tuned so that they are suitable for
measurement of specific petroleum products or group types. Examples of modified
GC-based methods include GRO, DRO, and TPHV methods. These modified
methods can be particularly useful when there is information on the source of cont-
amination, but method results should be interpreted with the clear understanding
that a modified method was used for detection of a specific carbon range. It is essen-
tial that the user understand what hydrocarbons a GC-based method can and cannot
detect and how results are quantified. For example, BTEX is a subset of TPHV. If
benzene, toluene, ethylbenzene, and the three xylene isomers are present in a
sample, they will be quantified along with the other TPHV components. The TPHV
measurement typically is greater than the sum of the BTEX measurements. Gasoline
should not be quantified by adding the TPHV and BTEX quantities together.
23

Interpretation of GC-based TPH data can be complicated and the analytical
method should always be considered when interpreting concentration data. A
volatile range TPH analysis may be very useful for quantifying TPH at a gasoline
release site, but a volatile range TPH analysis will not detect the presence of lube
oil. In addition, a modified GC-based method which has been specifically selected
for detection of gasoline-range organics at a gasoline-contaminated site may also
detect hydrocarbons from other petroleum releases because fuel carbon ranges
frequently overlap (See discussion in Section 3). Gasoline is found primarily in the
volatile range. Diesel fuel falls primarily in an extractable range. Jet fuel overlaps
both the volatile and semivolatile ranges. However, the detection of different kinds
of petroleum does not necessarily indicate that there have been multiple releases
at a site. Analyses of spilled waste oil will frequently detect the presence of gasoline,
and sometimes diesel. This does not necessarily indicate multiple spills. All waste
oils contain some fuel. As much as 10% of used motor oil can consist of gasoline
(Owen and Coley, 1990). The fuel gets into the oil as combustion chamber gases
blowing past the piston rings - a more pronounced problem in high mileage
engines with worn rings. Liquid fuel will also seep past rings under cold start and
warm-up conditions (Rhodes et al., 1994).
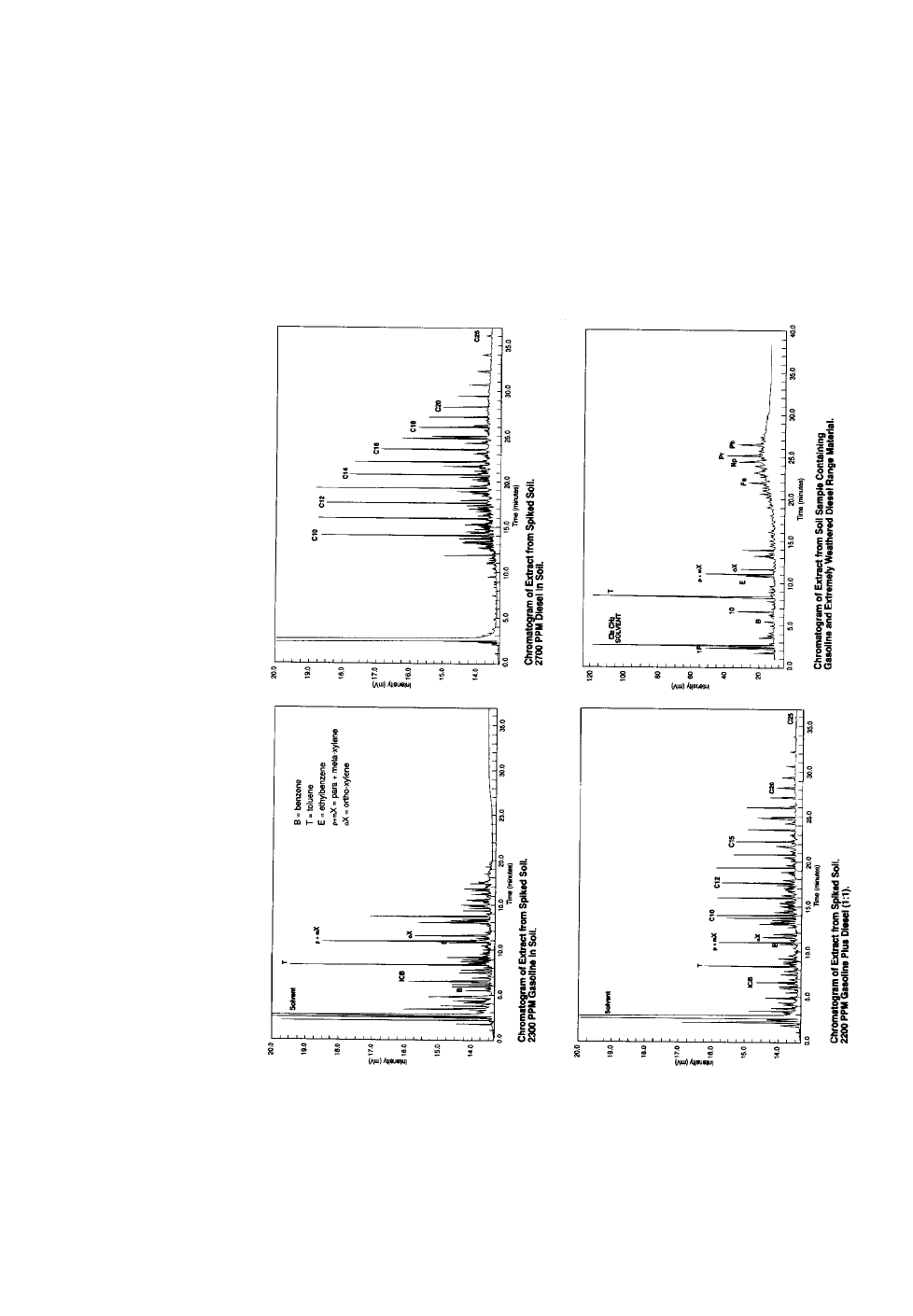
If the type of contaminant is unknown, a “fingerprint” analysis can help identify
it. A “fingerprint”, or pattern recognition, analysis is a direct injection GC/FID
analysis where the chromatogram is compared to chromatograms of reference
materials. Certain fuels can be identified by characteristic, reproducible chro-
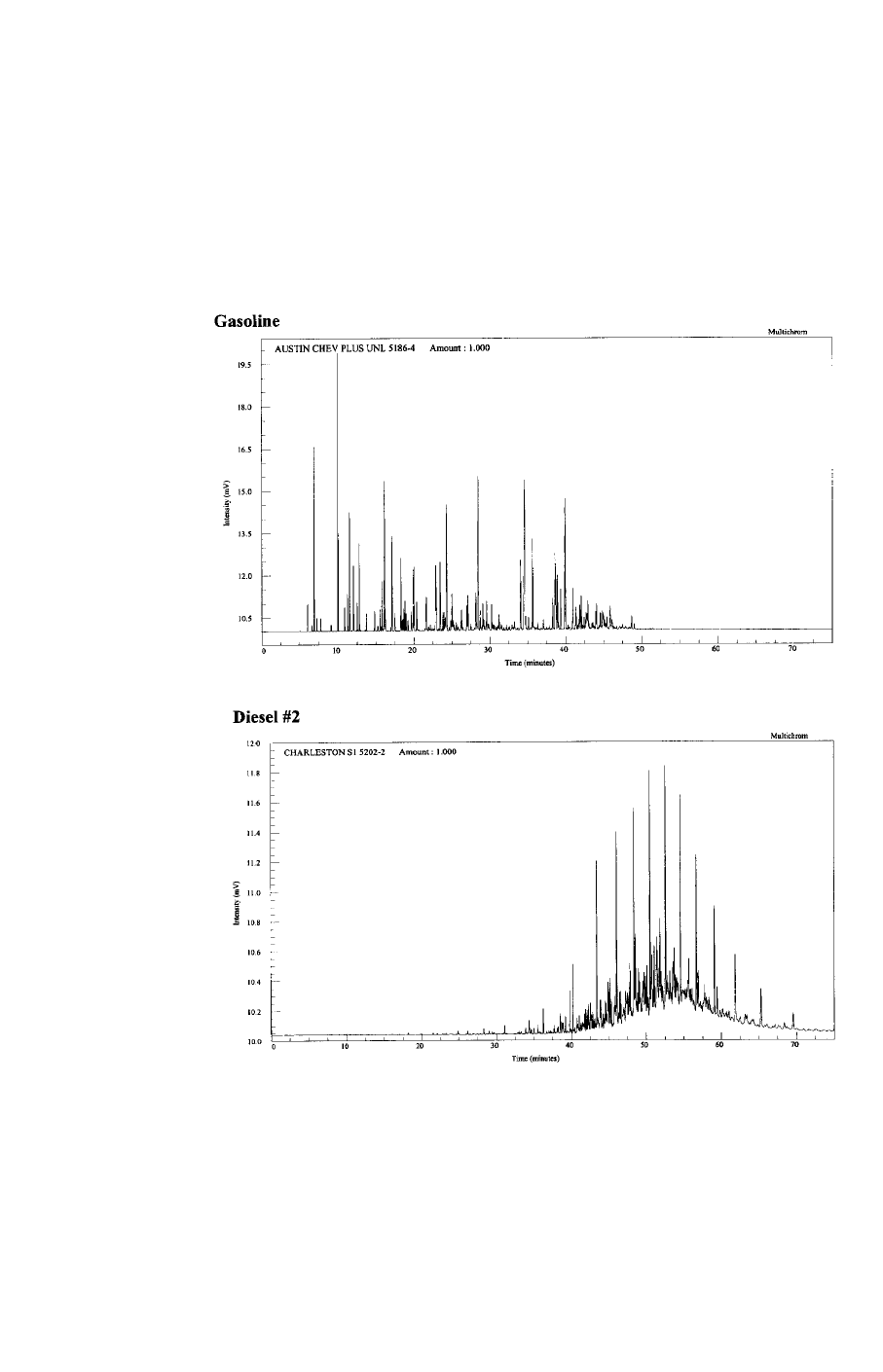
matographic patterns. For example, chromatograms of gasoline and diesel differ
considerably, as shown in Figures 3 and 4. There are complicating factors, however.
Many hydrocarbon streams may have similar fingerprints. Diesel #2 and #2 fuel oil
both have the same boiling point range and chromatographic fingerprint. A fin-
gerprint can be used to conclusively identify a mixture when a known sample of
that mixture or samples of the mixture’s source materials are available as refer-
ences. As a fuel evaporates or biodegrades, its pattern can change so radically that
identification becomes difficult. Consequently, a GC “fingerprint” is not a conclu-
sive diagnostic tool. These chromatograms must be interpreted by experienced
analysts. While GC-based TPH and pattern recognition methods are very similar,
TPH methods stress calibration and quality control, while pattern recognition
methods stress detail and comparability.
Interferences/Limitations
The GC-based methods usually cannot quantitatively detect compounds below C
6
because these compounds are highly volatile and interference can occur from the
solvent peak. As much as 25% of fresh gasoline can be below C
6
. This is not a
problem for the analyst with weathered gasoline range and/or diesel range conta-
mination because most of the very volatile hydrocarbons (<C
6
) may no longer be
present in the sample.
GC-based TPH methods may also have problems quantifying polar hydrocarbon
constituents (nitrogen, oxygen, and sulfur containing molecules). Some polar
hydrocarbon constituents are too reactive to pass through a gas chromatograph
and thus will not reach the detector for measurement.
24
25
Figure 3. Gas Chromatograms of Fuels.
26
Figure 4. GC “Fingerprint” Chromatograms of Soil Contamination.

Oxygenated gasolines are sometimes analyzed by GC-based methods. Efficiency
of purge methods is lower for oxygenates such as ethers and alcohols. GC detector
response to oxygenates is lower relative to hydrocarbons. Therefore TPH results
will tend to be biased slightly low for ether-containing fuels compared to equivalent
amounts of traditional gasolines. Methanol and ethanol elute before C
6
; conse-
quently, they are not quantified and may not even be detected due to coelution
with the solvent. TPH results for fuels containing these alcohols also will have a
negative bias.
GC-based methods may overestimate TPH concentrations due to the detection
of non-petroleum compounds. Chlorinated compounds may be detected by GC-
based methods and reported as TPH. While leaded gasolines contain a few parts
per million 1,2-dichloroethane and 1,2-dibromoethane as part of the lead additive
package, unleaded gasolines contain no chlorinated compounds. Solvents such as
tetrachloroethene and trichloroethane may come from dry cleaning operations,
parts degreasing, and semiconductor manufacturing. In addition, cleanup steps do
not perfectly separate petroleum hydrocarbons from biogenic material. Plant oils
and waxes are sometimes extracted from vegetation-rich soils and quantified by GC
methods as TPH. Silica gel cleanup may help to remove this interference but may
also remove some polar hydrocarbons.
Because petroleum is made up of so many isomers, many compounds, especial-
ly those above about C
8
, coelute with isomers of nearly the same boiling point.
These unresolved compounds are referred to as the unresolved complex mixture
(UCM). They are legitimately part of the petroleum signal, and unless state regu-
lations specify otherwise, should be quantified. Quantifying UCM requires a base-
line-to-baseline integration mode rather than a peak-to-peak integration mode, as
shown in Figure 5. Baseline to baseline integration quantifies all hydrocarbons in
a sample including the UCM, while in the peak-to-peak integration mode, only the
individual resolved hydrocarbons (not including the UCM) are quantified.
Laboratories rarely report integration modes; therefore, it is wise to ask what inte-
gration mode has been used. Some researchers argue that the UCM also may
contain some bacterial metabolites of petroleum hydrocarbons, generally alcohols,
aldehydes and acids. However, many of these polar compounds will not pass
through a chromatographic column and others can be removed with silica gel.
5.2 INFRARED SPECTROSCOPY (IR) TPH METHODS
For IR-based methods, TPH is defined as anything extractable by a solvent (Freon
113), which is not removed by silica gel and can be detected by IR at a specified
wavelength. The primary advantage of IR-based TPH methods is that they are
simple, quick and inexpensive. Detection limits for a commonly used IR-based TPH
method, EPA Method 418.1, are approximately 1 mg/L in water and 10 mg/kg in
soil. This TPH method often suffers from poor accuracy and precision, especially
for heterogeneous soil samples. IR-based methods give no information on the type
of fuel present, no information about the presence or absence of toxic molecules,
and no specific information about potential risk associated with the contamination.
27

28
Figure 5. Integration Modes.

Overview of the Technique
Infrared spectroscopy measures the vibration (stretching and bending) that occurs
when a molecule absorbs energy (heat) in the infrared region of the electromag-
netic spectrum. Different functional groups and bond types have different IR
absorption frequencies and intensities.
IR-based TPH methods measure the absorbance of the C-H bond. Most IR-based
methods in the United States typically measure the absorbance at a single fre-
quency (usually 2930 cm
-1
) which corresponds to the stretching of aliphatic CH
2
groups. Some methods, especially in Europe, use multiple frequencies including
2960 cm
-1
for CH
3
groups and 2900 to 3000 cm
-1
for aromatic C-H bonds.
Samples are extracted with a suitable solvent (i.e., a solvent with no C-H bonds).
Biogenic polar materials are removed with silica gel. Some polar petroleum hydro-
carbons may be lost in the silica gel cleanup. The absorbance of the silica gel eluate
is measured at the specified frequency and compared to the absorbance of a stan-
dard or standards of known petroleum hydrocarbon concentration. The IR
absorbance is a measurement of the sum of all the compounds contributing to the
TPH result. IR-based TPH methods cannot provide information on the type of
hydrocarbon contamination.
The extraction solvent for measuring TPH in soil must not contribute any C-H
stretching to the measurement. The most frequently specified solvent has been
Freon-113. This solvent is no longer being manufactured, in accordance with the
Montreal Protocol on Substances that Deplete the Ozone Layer and the Clean Air
Act Amendments of 1990. Some labs are depleting stockpiled Freon or redistilling
used Freon. Others have switched to alternate solvents. Carbon tetrachloride has
been used for IR-based methods (mostly in the European community). It is in
limited use in the U.S. because it is a known carcinogen and also affects the ozone
layer. Tetrachloroethene (also known as perchloroethylene, or PERC) is currently
being used by some U.S. labs. Solvents such as methanol, methylene chloride, or
hexane are not suitable for an IR-based method because they contain C-H bonds.
For all IR-based TPH methods, the C-H absorbance is quantified by comparing
it to the absorbance of standards of known concentration. An assumption is made
that the standard has an aliphatic-to-aromatic ratio and IR response similar to that
of the sample. Consequently, it is important to use a calibration standard as similar
to the type of contamination as possible. EPA Method 418.1 specifies a calibration
mixture of 15:15:10 n-hexadecane: isooctane: chlorobenzene.
Example Methods
The IR-TPH method that has been most frequently used is EPA Method 418.1
(EPA-600/4-79-020 Methods for Chemical Analysis of Water and Wastes). The
method is appropriate only for water samples. A separatory funnel liquid/liquid
extraction technique is used to extract the hydrocarbons from the water. Standard
Methods for the Examination of Water and Wastewater, 17th Ed., 1989, lists in Method
5520D a Soxhlet extraction technique for sludges. This extraction is frequently
used to adapt Method 418.1 to soil samples. Both EPA and Standard Methods specify
Freon 113 as the solvent of choice. At one time, the EPA issued a proposed Method
9073, an IR-based method with a Soxhlet extraction step suitable for sediment and
29

soil. This method was not included in update III of EPA SW-846, 3rd edition. The
EPA did issue Method 3560, an IR-based supercritical fluid extraction method for
diesel range contamination, which may become more popular as the extraction
technique gains maturity.
Method 418.1 results should not be expected to match extractable-range GC
results if the hydrocarbons being measured by IR have a wider boiling range than
those specified for the extractable TPH measurement. Method 418.1 is not limited
by carbon range, and will detect heavier hydrocarbons than GC-based TPH analy-
ses can measure. Furthermore, a positive bias has been reported for 418.1 vs.
8015M (GC-based) measurements in weathered limestones, silts, and clays caused
by ultra-fine particles in the extract (Thomey et al., 1989).
What Do IR Methods Measure?
At a frequency of 2930 cm
-1
, IR spectrometers measure the energy absorbance of
aliphatic C-H bonds. IR-based methods can measure any extracted compounds that
have alkyl groups (C-H groups) in the molecule. Any molecule that contains even
one C-H bond will contribute to the measurement.
Compounds have different IR responses. Single frequency IR methods will
measure straight chain paraffins, cycloalkanes, alkenes (IR response is lessened),
substituted aromatics (IR response is lessened), PAHs if they have alkyl groups on
them, and oxygenated molecules - ethers and alcohols (IR response is lessened).
IR-based methods using a single 2930 cm
-1
frequency will not adequately measure
benzene or naphthalene because they do not contain alkyl groups, but only aro-
matic C-H bonds.
Interferences/Limitations
Similar to GC-based TPH methods, IR-based TPH data must be interpreted after
considering certain limitations and interferences that can affect data quality. For
example, not all laboratories measure the C-H absorbance exactly the same way.
Within the set of methods that specify a single IR measurement, some methods call
for the measurement at precisely 2930 cm
-1
while others, including EPA Method
418.1, call for the measurement at the absorbance maximum nearest 2930 cm
-1
.
This variation can make a significant difference in the magnitude of the TPH
result, and can lead to confusion when comparing duplicate sample results from
laboratories that do not use the same protocol. If only C-H absorbance is mea-
sured, IR-based TPH methods will potentially underestimate TPH concentrations
in samples that contain petroleum constituents such as benzene and naphthalene
that do not contain alkyl C-H groups.
Because a TPH result is calculated as if the aromatics in the sample were present
in the same ratio as in the calibration standard, accuracy depends upon use of a
calibration standard as similar to the type of contamination as possible. Use of a
dissimilar standard will tend to create a positive bias in highly aliphatic samples and
a negative bias in highly aromatic samples. P.C. Hayes et.al. report that the stan-
dard Method 418.1 calibration mix will give higher results for diesel (predomi-
30
nantly aliphatic) than for gasoline (up to 50% aromatic). Some states use the same
hydrocarbon or hydrocarbon type, rather than the specified Method 418.1
mixture, as a calibration standard.
IR methods are also prone to interferences (positive bias) from nonpetroleum
sources. As shown in Figure 6, most organic compounds have some type of alkyl
group associated with them whether petroleum-derived or not.
5.3 GRAVIMETRIC TPH METHODS
Gravimetric methods measure anything extractable by a solvent, not removed during
solvent evaporation, and capable of being weighed. Some gravimetric methods
include a cleanup step to remove biogenic material. Those that do are considered
TPH methods. Those that do not are considered oil and grease (O&G) methods.
The advantage of gravimetric methods is that they are simple, quick, and inexpen-
sive. Detection limits are approximately 5-10 mg/L in water and 50 mg/kg in soils.
These methods are not especially suitable for measurement of light hydrocarbons that
volatilize at temperatures below 70-85°C. They are recommended for TPH measure-
ment only for very oily sludges, for samples containing heavy molecular weight hydro-
carbons, or for aqueous samples when hexane is preferred as the solvent.
Gravimetric methods give no information on the type of fuel present, no infor-
mation about the presence or absence of toxic compounds, and no specific infor-
mation about potential risk associated with the contamination.
31
Figure 6. Infrared Spectra from Petroleum and Nonpetroleum Sources.
Area of IR-TPH
measurement 2930cm
-1
3000 2800
cm
-1
Peat Moss
Wetland Soils
Gasoline
Wood Ash

Overview of the Technique
TPH compounds are extracted into a suitable solvent. Biogenic polar materials typ-
ically may be partially or completely removed with silica gel. The solvent is evapo-
rated and the residue is weighed. This quantity is called TPH or O&G and is report-
ed as a percent of the total soil sample dry weight. These methods are better suited
for heavy oils because they include an evaporation step.
Example Methods
There are a variety of gravimetric oil and grease methods suitable for testing water
and soil samples. These methods have been commonly used for National Pollutant
Discharge Elimination System (NPDES) permit measurements under the Clean
Water Act (e.g., EPA SW-846 9070 Total Recoverable Oil and Grease; EPA 413.1 Oil
and Grease, Total Recoverable).
One gravimetric method, EPA Method 9071, is recommended for measuring
TPH in oily sludges. Technically the result is an oil and grease result because no
cleanup step is used. “Method 9071 is used to recover low levels of oil and grease
by chemically drying a wet sludge sample and then extracting it using Soxhlet
apparatus. Results are reported on a dry-weight basis. Method 9071 is used when
relatively polar heavy petroleum fractions are present, or when the levels of non-
volatile greases challenge the solubility limit of the solvent. Specifically, Method
9071 is suitable for biological lipids, mineral hydrocarbons, and some industrial
wastewaters.” (EPA SW-846, Method 9071)
The EPA proposed Method 1664 as a “method for determination of n-hexane
extractable material… that is not adsorbed by silica gel in surface and saline waters
and industrial and domestic aqueous wastes (EPA, 1994).” This method provides
for the incorporation of a cleanup step. It has been designed to replace methods
based on Freon 113.
What Do Gravimetric Methods Measure?
Gravimetric “oil and grease” methods such as EPA SW-846 9071 measure anything
that dissolves in the solvent and remains after solvent evaporation. These sub-
stances include hydrocarbons, vegetable oils, animal fats, waxes, soaps, greases and
related biogenic material. Gravimetric TPH methods, such as proposed EPA
Method 1664, measure anything that dissolves in the solvent and remains after
silica gel treatment and solvent evaporation. All gravimetric methods measure any
suspended solids that are not filtered from solution, including bacterial degrada-
tion products and clay fines. Method 9071 specifies using cotton or glass wool as a
filter. Proposed EPA Method 1664 suggests using a 0.45-micron filter that removes
suspended solids.
Interferences/Limitations
Because extracts are heated to remove solvent, these methods are not suitable for
measurement of light hydrocarbons (i.e. less than C
15
) that volatilize at tempera-
tures below 70-85°C. Petroleum fuels, from gasoline through #2 fuel oils, are par-
32

tially lost in the solvent removal operation. In addition, soil results that are report-
ed on a dry-weight basis suffer from potential losses of light hydrocarbons during
moisture determination where the matrix is dried at approximately 103-105°C for
several hours in an oven.
Proposed Method 1664 uses n-hexane as a solvent. Hexane is not a good solvent
for high molecular weight petroleum compounds. In theory, proposed Method
1664 should yield lower results than TPH methods using Freon or chlorinated sol-
vents for samples contaminated with heavy oils. In practice, proposed Method 1664
has yielded results that are sometimes lower and sometimes higher than duplicate
Freon-extracted samples, suggesting that extraction efficiency depends upon a
variety of factors, not just solvent type.
Method 9071 currently uses Freon 113 as a solvent, but replacement solvents are
needed as Freon 113 use is terminated. Replacement solvents are limited only by
their ability to evaporate on a water bath at 70°C.
5.4 IMMUNOASSAY TPH METHODS
Immunoassay methods correlate TPH with the response of antibodies to specific
petroleum components. A number of different testing kits based on immunoassay
technology are available for rapid determination of TPH. The kits are self-contained
portable systems designed to conduct analytical work in the field. They include com-
ponents for sample preparation, instrumentation to read assay results, and
immunoassay reagents. Currently, most of these methods measure only aromatics.
Immunoassay is used as a screening technique because its precision and accuracy
are lower than standard laboratory methods such as GC/FID or IR. Immunoassay
measurements may be reported as a range or a single value. Typical detection limits
for TPH range from 10-500 mg/kg in soil and 200 to 500 µg/L in water.
Overview of the Technique
Antibodies are made of proteins that recognize and bind with foreign substances
(antigens) that invade host animals. Synthetic antibodies have been developed to
complex with petroleum constituents. The antibodies in the test kit are immobi-
lized on the walls of a special cell or filter membrane. Water samples are added
directly to the cell while soils must be extracted before analysis. A known amount
of labeled analyte is added after the sample. The label is typically an enzyme with
an affinity for the antibody. The sample analytes compete with the enzyme-labeled
analytes for sites on the antibodies. After equilibrium is established, the cell is
washed to remove any unreacted sample or labeled enzyme. Color development
reagents that react with the labeled enzyme are added. A solution that stops color
development is added at a specified time, and the optical density (color intensity)
is measured. Because the coloring agent reacts with the labeled enzyme, samples
with high optical density contain low concentrations of analytes. Concentration is
inversely proportional to optical density.
33

Example Methods
Test kits are available for TPH, BTEX, and PAH analysis (see discussion of
immunoassays in Section 6). The EPA included Method 4030, Petroleum
Hydrocarbons by Immunoassay, in Update III of SW-846.
What Do Immunoassay Methods Measure?
The antibodies used in immunoassay kits are generally designed to bond with
selected compounds such as BTEX and PAHs. A correction factor supplied by the
manufacturer must be used to calculate the TPH concentration. The correction
factor can vary depending on product type because it attempts to correlate total
TPH with the measured surrogates.
Interferences/Limitations
Immunoassay tests do not identify specific fuel types. They are best used as
screening tools.
Immunoassay tests are dependent on soil type and homogeneity. In particular,
for clay and other cohesive soils, the tests are limited by a low capacity to extract
hydrocarbons from the sample.
6. PETROLEUM GROUP TYPE MEASUREMENT: DETAILED REVIEW OF
SELECTED ANALYTICAL METHODS
This section describes group-type methods for petroleum hydrocarbons. Group-
type methods are designed to separate hydrocarbons into categories, such as satu-
rates, aromatics, and polars/resins, or “PIANO” - paraffins, isoparaffins, naph-
thenes, olefins, and aromatics. These High Performance Liquid Chromatography
(HPLC) and GC methods were typically developed for monitoring refinery
processes or evaluating organic synthesis products, although they are beginning to
be used for environmental applications. Column chromatographic methods that
separate saturates from aromatics are often used as preparative steps for further
analysis by GC/MS. Thin layer chromatography is sometimes used as a screening
technique for petroleum product identification.
6.1 THIN LAYER CHROMATOGRAPHY (TLC) GROUP TYPE METHODS
TLC is perhaps an underutilized test method in the environmental field. Organic
chemists sometimes use TLC to determine which stationary and mobile phases
provide the best separations. The same stationary and mobile phases may then be
used for more sophisticated HPLC analysis.
In the environmental field, TLC is best used for screening analyses and character-
ization of semivolatile and nonvolatile petroleum products. Precision and accuracy
of the technique is inferior to Method 8015 or 418.1 analysis, but when speed and
simplicity are desired, TLC may be a suitable alternative. For characterizations of
heavy petroleum products such as tar or asphalt, TLC has the advantage of separat-
ing compounds that are too heavy to pass through a GC. While TLC does not have
the resolving power of a GC, it is able to separate different classes of compounds.
34

Overview of the Technique
Thin layer chromatography analysis is fairly simple. Since TLC does not give highly
accurate or precise results, there is no need to perform the highest quality extrac-
tions. Soils are easily extracted by shaking or vortexing with solvent. Water samples
are extracted by shaking in a separatory funnel. If one suspects that interfering
compounds are present, silica gel can be added to clean the extract.
Sample extract aliquots are placed close to the bottom of a glass plate coated
with a stationary phase. The most widely used stationary phases are made of an
organic hydrocarbon moiety bonded to a silica backbone. For the analysis of petro-
leum hydrocarbons, a moderately polar material stationary phase works well.
The plate is placed in a sealed chamber with a solvent (mobile phase). The
solvent travels up the plate carrying compounds present in the sample. The dis-
tance a compound travels is a function of the affinity of the compound to the sta-
tionary phase relative to the mobile phase. Compounds with chemical structures
and polarities similar to the solvent travel well in the mobile phase. For example,
the saturated hydrocarbons seen in diesel fuel travel readily up a TLC plate in a
hexane mobile phase. Polar compounds such as ketones or alcohols travel a
smaller distance in hexane than saturated hydrocarbons.
After a TLC plate has been exposed to the mobile phase solvent for the required
time, the compounds present can be viewed by several methods. Polynuclear aro-
matic compounds (PAHs), other compounds with conjugated systems, and com-
pounds containing heteroatoms (nitrogen, oxygen, or sulfur) can be viewed with
longwave and shortwave UV light. The unaided eye can see other material. Also,
plates can be developed in iodine. Iodine has an affinity for most petroleum com-
pounds, including the saturated hydrocarbons, and stains the compounds a
reddish/brown color. Schematic examples of different products analyzed by TLC
are shown in Figure 7.
Example Methods
There is no current EPA Method using a TLC technique. The TLC approach is
considered to be a qualitative but useful tool for rapid sample screening.
What Do TLC Methods Measure?
TLC methods are usually qualitative. The analyst identifies the product present in
a sample by comparison with concurrently run standards of different petroleum
products. If the type of contamination is known, standards of known concentration
can be run alongside the sample to allow visual approximation of concentration.
An instrument called a densitometer can be used to measure sample concentra-
tion, but this approach removes two attractive features of TLC: cost and simplicity.
Interferences/Limitations
Limitations of TLC center on its moderate reproducibility, detection limits, and
resolving capabilities. Variability between operators can be as high as 30%.
Detection limits (without any concentration of the sample extract) are near 50
35
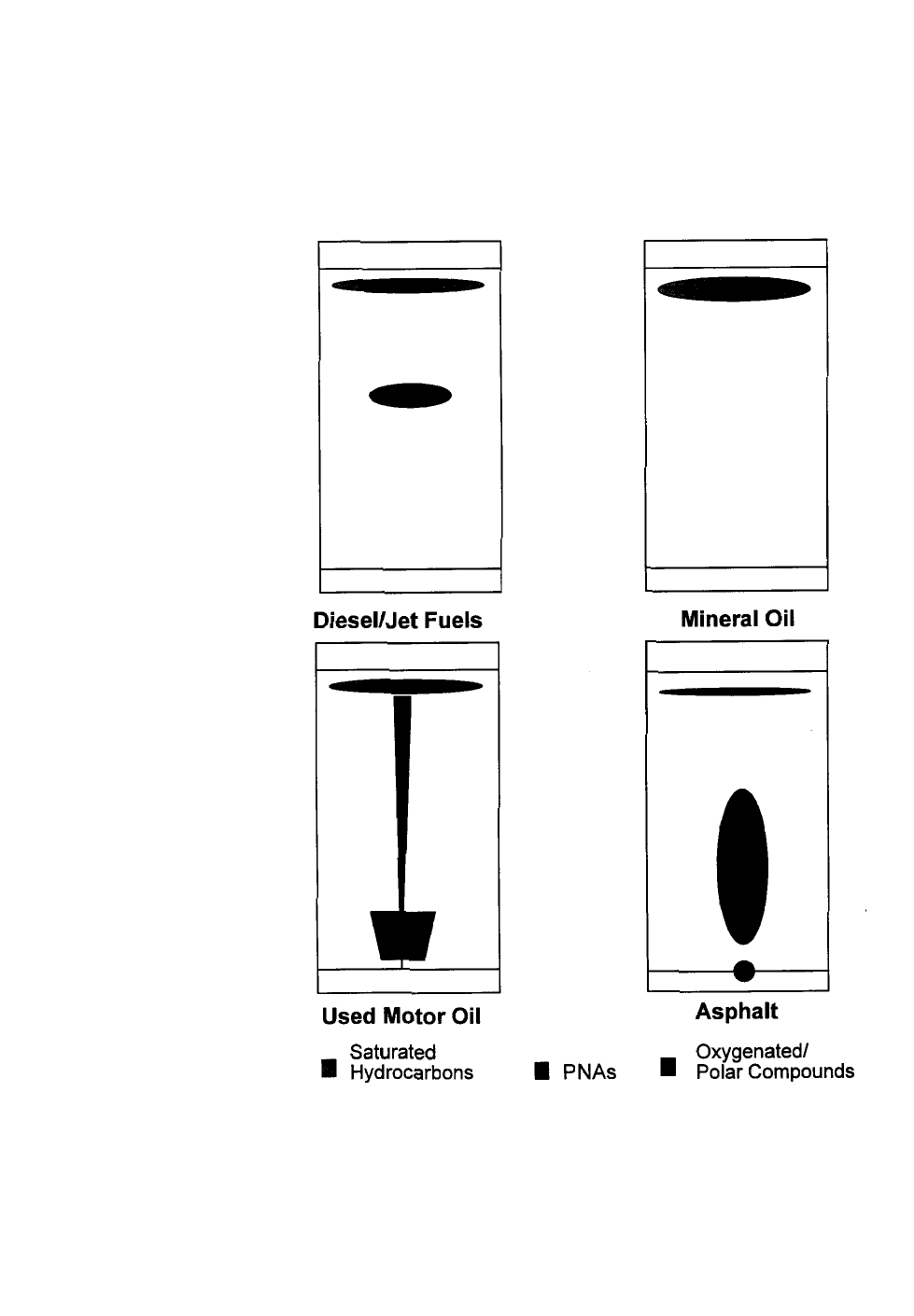
36
Figure 7. Schematic TLC Plates of Petroleum Products
(Plates Developed in Hexane).

ppm (mg/kg) for most petroleum products in soils. When the aromatic content of
a material is high, as with bunker C, the detection limit can be near 100 ppm. It is
often not possible to distinguish between similar products such as diesel and jet
fuel. As with all chemical analyses, quality assurance tests should be run to verify
the accuracy and precision of the method.
6.2 IMMUNOASSAY GROUP TYPE METHODS
A number of different testing kits based on immunoassay technology are available
for rapid field determination of certain groups of compounds such as BTEX or
PAHs. These immunoassay screening kits are self-contained portable field kits that
include components for sample preparation, instrumentation to read assay results,
and immunoassay reagents.
Immunoassay is used as a screening technique because its precision and accura-
cy is lower than standard laboratory methods such as GC/FID or IR. Typical detec-
tion limits for BTEX are 2 mg/kg in soil and 200 µg/L in water. Typical detection
limits for PAHs are 1 mg/kg in soil and 10 µg/L in water.
Overview of the Technique
The immunoassay technique has been previously discussed (see Section 5.4).
Briefly, immunoassay tests measure TPH through the binding of petroleum con-
stituents to protein antibodies.
Example Methods
Test kits are available for BTEX, PAH and TPH measurement. The EPA has includ-
ed Method 4035, Polycyclic Aromatic Hydrocarbons by Immunoassay, in Update III
of SW-846.
What Do Immunoassay Methods Measure?
The antibodies used in immunoassay kits are designed to bond with specific com-
pounds such as monaromatics, (e.g., BTEX), and PAHs.
Interferences/Limitations
Immunoassay kits may display strong biases. Most of the BTEX kits have a low affin-
ity for benzene relative to toluene, ethylbenzene, xylenes, and other aromatic com-
pounds. The kits with low benzene affinity underestimate the actual benzene levels.
Since benzene is often the dominant compound in leachates due to its high solu-
bility, a low sensitivity for benzene is undesirable. Benzene-specific kits are being
developed. In general, test kit specificity varies by manufacturer.
The quality of PAH analysis is often dependent on the extraction efficiency. Clay
and other cohesive soils lower the ability to extract PAHs. Another potential
problem with PAH analysis is that the test kits may have different responses for dif-
ferent PAH compounds.
37

In addition, the target compound test kits are often subject to false positives
because the antibodies have some affinity for nontarget compounds. In general,
test kits give less than 25% false positives and less than 5% false negatives.
7. INDIVIDUAL PETROLEUM CONSTITUENT MEASUREMENT:
DETAILED REVIEW OF SELECTED ANALYTICAL METHODS
Many common environmental methods measure individual petroleum con-
stituents or “target compounds” rather than the whole TPH signal. Each method
measures a suite of compounds selected because of their toxicity and common use
in industry. For organic compounds, the EPA has generated three series of target
compound methods:
• EPA 500 series, “Organic Compounds in Drinking Water,” as regulated
under the Safe Drinking Water Act.
• EPA 600 series, “Methods for Organic Chemical Analysis of Municipal and
Industrial Wastewater,” as regulated under the Clean Water Act.
• EPA SW-846 series, “Test Methods for Evaluating Solid Waste:
Physical/Chemical Methods,” as promulgated by the USEPA, Office of
Solid Waste and Emergency Response.
The 500 and 600 series methods provide parameters and conditions for the analy-
sis of drinking water and wastewater, respectively. EPA SW-846 is geared toward the
analysis of nearly all matrices including industrial wastes, soils, sludges, sediments,
and water miscible and nonwater miscible wastes. It also provides for the analysis of
groundwater and wastewater but is not used to evaluate compliance of public drink-
ing water systems. Many of these EPA methods are capable of detecting the same
target analytes; however, they often differ in applicability, sample preparation,
instrument operating conditions, quality assurance calibration requirements,
method detection limits, quality control measures, and reporting requirements.
Selection of one method over another is often dictated by the nature of the sample
and the particular compliance or cleanup program for which the sample is being
analyzed. It is essential to recognize that capabilities and requirements vary between
methods when requesting any analytical method or suite of methods.
Table 5 lists the most common EPA methods used to detect and measure con-
stituents of petroleum hydrocarbons. For some of the methods, additional target
compounds not specified in the method may be added upon validation. Since it is
not possible to list every method or identify method requirements and compo-
nents in a “method comparison,” the reader is advised to refer to the appropriate
EPA series or method for more information or to consult with an analytical labo-
ratory manager for additional guidance.
Most compound-specific methods use a GC/selective detector, HPLC, or
GC/MS technique. GC and HPLC techniques identify analytes based on their
retention times and can be subject to interferences. For this reason, these tech-
niques involve use of selective detectors. GC techniques typically require second-
column confirmation. GC/MS provides confirmation of the identity of an analyte
through its retention time and unique spectral pattern.
38
39
TABLE 5. Cross Reference to USEPA Target Analyte Methods
USEPA Series/Methods 500 Series 600 Series SW-846 Methods
Target Analytes 502.2 524.2 602 610 624 625 8015 8020 8021 8100 8240 8260 8270 8310
Acenaphthene • • • • •
Acenaphthylene • • • • •
Anthracene • • • • •
Benzene • • • • • • • • •
Benz(a)anthracene • • • • •
Benzo(a)pyrene • • • • •
Benzo(b)fluoranthene • • • • •
Benzo(ghi)perylene • • • • •
Benzo(j)fluoranthene •
Benzo(k)fluoranthene • • • • •
Chrysene • • • • •
Dibenz(a,h)anthracene •
Dibenz(a,h)acridine •
Dibenz(a,j)acridine ••
7H-Dibenzo•c,g)carbazole •
Dibenzo•a,h)anthracene • • • • •
Dibenzo•a,e)pyrene ••
Dibenzo•a,h)pyrene •
Dibenzo•a,i)pyrene •
7,12-Dimethylbenz•a)anthracene •
Ethylbenzene • • • •
◆
•• ••
Fluoranthene • • • • •
Fluorene • • • • •
Indeno•1,2,3-cd)pyrene • • • • •
3-Methylcholanthrene ••
40
TABLE 5. (Continued)
USEPA Series/Methods 500 Series 600 Series SW-846 Methods
Target Analytes 502.2 524.2 602 610 624 625 8015 8020 8021 8100 8240 8260 8270 8310
2-Methylnaphthalene •
Methyl-t-butyl ether •MTBE) • •
Naphthalene • • • • • • • • •
Phenanthrene • • • • •
Propylbenzene • • •
Pyrene • • • •
Styrene • •••
1,2,4-Trimethylbenzene • • • •
1,3,5-Trimethylbenzene • • • •
Toluene • • • •
◆
•• ••
o-Xylene • •
◆◆◆
•• ••
m-Xylene • •
◆◆◆
•• ••
p-Xylene • •
◆◆◆
•• ••
• = Compounds specified in the method.
◆
= Compounds not specified in the method, but may be target compounds under the methodís general or modified.

Compound-specific methods frequently require samples to be diluted to keep
the most concentrated analytes within the calibration range, or to protect the
detectors from difficult-to-remove contamination. These requirements often cause
low concentration analytes to fall below detection limits. Remedies for this
problem may include analyzing the sample at more than one concentration or
choosing a detection method with a wider dynamic range. In some cases, the detec-
tion limit problem cannot be resolved without risking potential contamination of
the instrumentation.
7.1 GAS CHROMATOGRAPHY WITH PHOTOIONIZATION DETECTION (GC/PID) PETROLE-
UM CONSTITUENT METHODS
The photoionization detector (PID) can be tuned to be selective to aromatics. The
advantage of GC/PID is its selectivity and sensitivity. Typical detection limits for
light aromatics are 0.5 µg/L in water and 5 µg/kg in soil.
In some method variations, the GC is equipped with two detectors in series. A PID
is used to measure benzene, toluene, ethylbenzene, and the xylenes (BTEX), while
an FID measures the total TPH signal (which includes the BTEX compounds).
Overview of Technique
For volatiles, the gas chromatograph is generally interfaced with a purge and trap
system as described in the section on GC-Based Methods. The photoionization
detector works by bombarding compounds with ultraviolet (UV) light, generating
a current of ions. Compounds with double carbon bonds, conjugated systems
(multiple carbon double bonds arranged in a specific manner), and aromatic rings
are easily ionized with the UV light generated by the PID lamp, while most satu-
rated compounds require higher energy radiation.
Example Methods
EPA Method 8020 targets volatile aromatics. This method is often referred to as
BTEX analysis, though the method includes other volatile aromatics in addition to
BTEX. The method is similar to most volatile organic GC methods. Sample prepa-
ration and introduction is typically by EPA Method 5030 (Purge and Trap Analysis).
What Do GC/PID Methods Measure?
A PID measures volatile aromatic compounds, such as benzene, toluene, ethylben-
zene and the xylenes. Some oxygenates such as methyl-t-butyl ether (MTBE) are
detected. Additionally, many olefins and some branched alkanes and cycloalkanes
are detected.
Interferences/Limitations
In all GC analyses, compounds are identified by the retention time of the analyte.
Although the selectivity of the PID helps to minimize false positives, GC columns
must be selected carefully to minimize potential interferences. Second
41

column/second detector confirmation should be used to increase reliability of
identification. In highly contaminated samples where the PID is responding to
large amounts of material, running a volatile GC/MS analysis (EPA Method 8240
or 8260) for confirmation can be beneficial.
Certain false positives are common with EPA Method 8020. Trimethylbenzenes,
and gasoline constituents are frequently identified as chlorobenzenes by Methods
602 and 8020 because these compounds elute with nearly the same retention times
from nonpolar columns. Cyclohexane is often mistaken for benzene by combined
Method 8015/8020 analyses, because both compounds are detected by a 10.2 eV
PID detector and have nearly the same elution time from a Method 8015 nonpo-
lar column. The two compounds have very different retention times on a more
polar column like the type specified by the Method 8020, but a more polar column
skews the carbon ranges measured by Method 8015. False positives for oxygenates
in gasoline are common, especially in highly contaminated samples.
Samples containing distillate and heavier hydrocarbons are generally not ana-
lyzed by GC/PID. The PID detector is easily contaminated and desensitized by the
heavier compounds.
7.2 GAS CHROMATOGRAPHY WITH FLAME IONIZATION DETECTION (GC/FID) PETROLEUM
CONSTITUENT METHODS
The flame ionization detector (FID) is a nonselective detector that employs a hydro-
gen-fueled flame to ionize organic compounds. The advantage of GC/FID is its sen-
sitivity to a broad range of hydrocarbon compounds. GC/FID systems are some-
times used to measure PAHs, and typical detection limits for PAHs are 10 µg/L in
water and 330 µg/kg in soil.
Flame ionization detectors are destructive detection systems because the analyte
is combusted during detection. They may only be used as the second detector in
cases where a GC is equipped with two detectors in series. In some methods, an FID
may be used to measure the total TPH signal (as in a modified EPA Method 8015)
after a PID has been used to measure light aromatics such as benzene, toluene, eth-
ylbenzene, and the xylenes (BTEX).
Overview of Technique
For semivolatiles, the gas chromatograph is generally equipped with either a packed
or capillary column. Either neat or diluted organic liquids can be analyzed via direct
injection, and compounds are separated during movement down the column.
The FID uses a hydrogen-fueled flame to ionize compounds that reach the detector.
Any compound that burns can be detected by an FID. Ionized molecules produce a
current proportional to the total volatile organic vapor concentration in the sample, and
this current change is recorded as a signal. Calibration standards can be used to quan-
tify instrument response, and analyte signals can thus be converted to concentrations.
Example Methods
EPA Method 8100 targets PAHs. Injection of sample extracts directly onto the
column is the preferred method for sample introduction for this packed-column
42

GC technique. Detection limits for PAHs are not specified in the method, but
quality control acceptance criteria values range from 5-100 µg/L.
What Do GC/FID Methods Measure?
A GC/FID system can be used for the separation and detection of nonpolar
organic compounds. Semivolatiles such as PAHs are among the analytes that can
be readily resolved and detected using a GC/FID system. If a packed GC column is
used, four pairs of compounds may not be adequately resolved and are reported as
a quantitative sum: anthracene and phenanthrene, chrysene and
benzo(a)anthracene, benzo(b)fluoranthene and benzo(k)fluoranthene, and
dibenzo(a,h)anthracene and indeno(1,2,3-cd)pyrene. This problem can be
addressed through the use of a capillary column in place of a packed column. An
FID can also detect various interfering compounds, including sulfur compounds.
Other co-extracted compounds such as phthalates, which are common environ-
mental contaminants due to their use in plastics, can also interfere with PAH analy-
sis by GC/FID.
Interferences/Limitations
In all GC analyses, compounds are identified by the retention time of the analyte.
Therefore, GC columns must be selected carefully to minimize potential interfer-
ences. FIDs are nonselective detectors and thus are sensitive to interfering com-
pounds as well as analytes. Interferences can hide analyte signals and thus necessi-
tate a sample cleanup step for proper quantification and identification of analytes
using GC/FID.
Second column/second detector confirmation should be used to increase relia-
bility of identification. In highly contaminated samples where the FID is respond-
ing to large amounts of material, running a semivolatile GC/MS analysis for con-
firmation can be beneficial.
7.3 HIGH PERFORMANCE LIQUID CHROMATOGRAPHY (HPLC) PETROLEUM CON-
STITUENT METHODS
An HPLC system is used to measure concentrations of target semivolatile and non-
volatile petroleum constituents. Unlike GC systems that require complete volatiliza-
tion of the sample so that it can then pass into the chromatograph, LC systems only
require that the sample be dissolved in a solvent compatible with those used in the
separation. The HPLC detector most often used in petroleum environmental
analysis is the fluorescence detector. These detectors are particularly sensitive to
aromatic molecules, especially the PAHs. A UV detector may be used to measure
compounds which do not fluoresce.
Method detection limits for PAHs by HPLC, EPA Method 8310, range from
0.013-2.3 µg/L.
43

Overview of Technique
PAHs are extracted from the sample matrix with a suitable solvent, which is then
injected into the HPLC. Usually the extract must be filtered because fine particulate
matter can collect on the inlet frit of the HPLC column, resulting in high back-pres-
sures and eventual plugging of the column. For most hydrocarbon analyses, reverse
phase HPLC (i.e., using a nonpolar column packing with a more polar mobile
phase) is used. The most common bonded phase is the octadecyl or C
18
phase. The
mobile phase is commonly aqueous mixtures of either acetonitrile or methanol.
After the chromatographic separation, the analytes flow through the cell of the
detector. A fluorescence detector shines light of a particular wavelength (the exci-
tation wavelength) into the cell. Fluorescent compounds absorb light and reemit
light of other, higher wavelengths (emission wavelengths). The emission wave-
lengths of a molecule are mainly determined by its structure. For PAHs, the emis-
sion wavelengths are mainly determined by the arrangement of the rings and vary
greatly between isomers. Some EPA methods specify the use of a filter-based detec-
tor, which measures all light above a certain wavelength. Many modern detectors
can tune excitation and emission wavelengths to maximize sensitivity and/or selec-
tivity for each analyte during a chromatographic run. At the very low concentra-
tions often found in environmental samples, fluorescence detection is linear and
requires the usual generation of calibration curves. Compounds are identified
based on their retention times.
A UV (ultraviolet) detector may be used for compounds like acenaphthylene,
which does not fluoresce. This detector measures the absorbance of ultraviolet
light by the analyte, although it is less sensitive than the fluorescence detector.
Example Methods
EPA Method 8310 targets 16 PAHs. Some of these PAHs, such as phenanthrene,
pyrene, and benzo(g,h,i)perylene, are commonly seen in products boiling in the
middle to heavy distillate range. The method uses an octadecyl column and an
aqueous acetonitrile mobile phase. Analytes are excited at 280 nm and detected at
emission wavelengths of >389 nm. Naphthalene, acenaphthene, and fluorene must
be detected by a less-sensitive UV detector because they emit light at wavelengths
below 389 nm. Acenaphthylene is also detected by UV detector.
What Do HPLC Methods Measure?
HPLC methods using fluorescence detection will measure any compounds that
elute in the appropriate retention time range and which fluoresce at the targeted
emission wavelength(s). In the case of Method 8310, the excitation wavelength
excites most aromatic compounds. These include the target PAHs but also many
derivatized aromatics, such as alkylaromatics, phenols, anilines, and heterocyclic
aromatic compounds containing the pyrrole (indole, carbazole, etc.), pyridine
(quinoline, acridine, etc.), furan (benzofuran, naphthofuran, etc.), and thiophene
(benzothiophene, naphthothiophene, etc.) structures. In petroleum samples, alkyl
PAHs are strong interfering compounds. For example, there are five
methylphenanthrenes and over 20 dimethylphenanthrenes. The alkyl substitution
44

does not significantly affect either the wavelengths or intensity of the phenan-
threne fluorescence. For a very long time after the retention time of phenan-
threne, the alkylphenanthrenes will interfere, affecting the measurements of all
later-eluting target PAHs.
Interferences/Limitations
Interfering compounds will vary considerably from source to source. Samples may
require a variety of cleanup steps to reach required method detection limits.
The emission wavelengths used by Method 8310 are not optimal for sensitivity of
the small ring compounds. With modern electronically-controlled monochroma-
tors, wavelength programs can be used which tune excitation and emission wave-
lengths to maximize sensitivity and/or selectivity for a specific analyte in its reten-
tion time window.
7.4 GAS CHROMATOGRAPHY WITH MASS SPECTROMETRY DETECTION (GC/MS)
PETROLEUM CONSTITUENT METHODS
A GC/MS system is used to measure concentrations of target volatile and semi-
volatile petroleum constituents. It is not typically used to measure TPH. The advan-
tage of GC/MS is the high selectivity, or ability to confirm compound identity
through retention time and unique spectral pattern. Because of the complexity of
their operation and of the interpretation of their output, GC/MS techniques tend
to be more costly than other GC techniques (GC/PID, GC/FID).
For volatile analytes, detection limits are as low as 1-5 µg/L for water and 20
µg/kg for soil. For semivolatile analytes, detection limits are as low as 5 µg/L for
water and 50 µg/kg for soil. A low-level volatiles method for soil is available, and 5
µg/kg detection limits can sometimes be achieved.
Overview of Technique
Mass spectrometer systems are designed to ionize compounds and scan for ions of
specific mass-to-charge ratios. Each compound breaks apart into a consistent, rec-
ognizable pattern of fragment ions. Ideally, coupling a gas chromatograph (GC)
with a mass spectrometer (MS) allows one to separate a mixture into its con-
stituents, ionize each constituent in turn, and identify the constituent compounds
by their fragmentation patterns.
Typical GC/MS outputs are shown in Figure 8. Each fragment ion is measured
as a ratio of its mass to charge. A scan of all the mass-to-charge ratios done in a frac-
tion of a second is known as a spectrum (Figure 8a). Every compound, with the
exception of isomers, has a unique spectrum, although the differences may be
subtle. A plot of the intensity of a single ion over time is called an ion chro-
matogram. Ion chromatograms can be used to identify trace concentrations of spe-
cific types of compounds in the presence of a complex mixture (Figure 8b). If all
the ion responses in a scan are summed, a total ion chromatogram can be gener-
ated (Figure 8c). The total ion chromatogram of a petroleum product is similar to
a GC/FID trace.
45
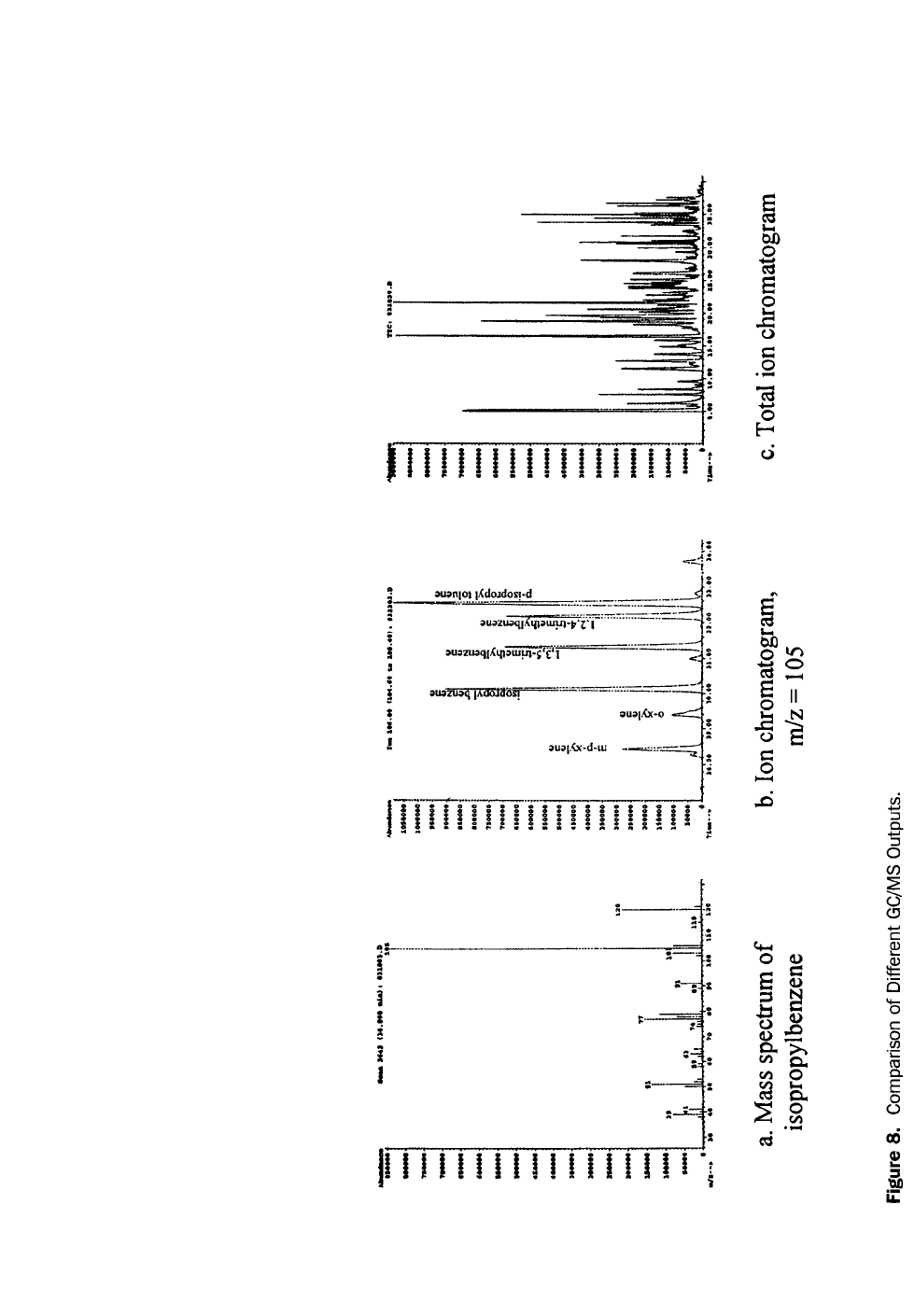
46
Figure 8. Comparison of Different GC/MS Outputs.

In a technique called selected ion monitoring, or SIM, the GC/MS is set up to
measure only selected target masses rather than scanning the full mass range. The
output looks like Figure 8b. This technique yields lower detection limits for specif-
ic compounds. At the same time, it gives the more complete information available
from the total ion chromatogram and the full-mass-range spectrum of each com-
pound. The technique is sometimes used to quantify compounds present at very
low concentrations in a complex hydrocarbon matrix. It can be used if the target
compound’s spectrum has a prominent fragment ion at a mass that distinguishes
it from the rest of the hydrocarbon compounds.
Example Methods
The current SW-846 GC/MS method for the analysis of volatile compounds is
Method 8260. A comparison with recently discontinued Method 8240 reveals that
most of the compounds listed in these methods are not typically found in petrole-
um products. Both methods measure some petroleum compounds, including
BTEX, ethylene dibromide and ethylene dichloride, which are additives associated
with leaded gasolines. C
3
Benzenes, some C
4
benzenes, and napthalenes (com-
pounds common to petroleum products) are also measured by Method 8260.
The most common method for GC/MS analysis of semivolatile compounds is
EPA SW-846 8270. The 8270 list includes 16 polycyclic aromatic compounds
(PAHs). Some of these PAHs are commonly seen in middle distillate to heavy
petroleum products. The method also quantifies phenols and cresols, compounds
that are not hydrocarbons but may occur in petroleum products. Phenols and
cresols are more likely found in crude oils and weathered petroleum products.
What Do GC/MS Methods Measure?
GC/MS methods identify compounds by retention time and mass spectrum. To
reduce the possibility of false positives, the intensities of one to three selected ions
are compared to the intensity of a unique target ion of the same spectrum. The
sample ratios are compared to the ratios of a standard. If the sample ratios fall
within a certain range of the standard, and the retention time matches the stan-
dard within specifications, the analyte is considered present (Figure 8).
Quantification is performed by integrating the response of the target ion only.
Interferences/Limitations
Mass spectrometers are among the most selective detectors, but they are still sus-
ceptible to interferences. Isomers have identical spectra, while many other com-
pounds have similar mass spectra. Heavy petroleum products can contain thou-
sands of major components that are not resolved by the gas chromatograph. As a
result, multiple compounds are simultaneously entering the mass spectrometer.
Different compounds may share many of the same ions, confusing the identifica-
tion process. The probability of misidentification is high in complex mixtures such
as petroleum products.
47

Sometimes, identification of compounds is accomplished by an automated
library search program. Because computer libraries can never contain all possible
petroleum isomers and frequently contain pesticides, plastics and other com-
pounds not found in petroleum, they frequently misidentify compounds.
Computer library identifications should only be used with extreme caution.
GC/MS is not a suitable technique for quantifying TPH. The response of an FID
is proportional to the mass of hydrocarbon present and is insensitive to the type of
hydrocarbon (e.g., aromatic, n-alkane and olefin). A mass spectrometer, however,
may have very different responses for two different hydrocarbon compounds. Since
petroleum products are complex mixtures of hydrocarbons, the same mass of two
different products may have two different responses on a mass spectrometer.
8. EVOLVING METHODS FOR PETROLEUM HYDROCARBON FRACTIONS
New petroleum analytical methods are being developed and used that identify and
quantify TPH as hydrocarbon fractions. Rather than quantifying a complex TPH
mixture as a single number, petroleum hydrocarbon fraction methods break the
TPH mixture into discrete hydrocarbon fractions, thus providing data that can be
used in a risk assessment and in characterizing product type and compositional
changes (e.g., weathering).
8.1 WHAT DO PETROLEUM FRACTION METHODS MEASURE?
TPH fraction methods can be used to measure both volatile and extractable hydro-
carbons. The currently available TPH fraction methods, including the TPHCWG
Method and the Massachusetts Department of Environmental Protection (MA
DEP) EPH/VPH Method, are most appropriate for measurement of hydrocarbons
in the approximate carbon range C
6
to C
28
. Lighter and heavier hydrocarbons may
have lower recoveries, but their detection is possible and may be enhanced by
method modification. MA DEP was the first to use a petroleum fraction approach
in characterizing and evaluating potential human health risk associated with petro-
leum contaminated sites. The MA DEP fractions are divided according to the
expected toxicity of individual constituents. The Working Group fractions are
based on the expected environmental behavior of individual petroleum con-
stituents. This grouping simplifies the environmental modeling needed to assess
potential human exposure to petroleum contamination. In both the MA DEP and
Working Group approaches, petroleum constituents are first divided into aliphatic
and aromatic compound fractions prior to further subdivision according to chem-
ical class and boiling ranges.
8.2 WHY USE PETROLEUM FRACTION METHODS?
In contrast to traditional TPH methods that report a single concentration number
for complex TPH mixtures, TPH fraction methods report separate concentrations
for discrete aliphatic and aromatic fractions. The available petroleum fraction
methods are GC-based and are thus sensitive to a broad range of hydrocarbons.
48

Identification and quantification of aliphatic and aromatic fractions allows one to
identify petroleum products and evaluate the extent of product weathering. These
fraction data also can be used in risk assessment. Together, the fractions represent
the mass of petroleum used to evaluate noncancer risk. Cancer risk is evaluated
separately from the fractions, using concentration data for individual carcinogenic
petroleum constituents (e.g., benzene and several PAHs).
8.3 EXAMPLES OF PETROLEUM FRACTION METHODS
Several TPH fraction methods are either in development and/or are gaining
regular usage. Two such methods include the TPHCWG Method and the MA DEP
EPH/VPH Method. The TPHCWG Method was developed with the assistance of
Shell Development Company. The Massachusetts EPH/VPH is the promulgated
TPH methodology for site cleanups in Massachusetts. The transition to this
method has occurred over several years, and state cleanup standards for both soil
and groundwater have been developed for the measured EPH/VPH fractions.
Petroleum fractions quantified in both of these methods have been paired with
toxicity criteria so that human health risk associated with exposures to petroleum-
contaminated media can be assessed.
TPHCWG Analytical Methodology
This method is designed to characterize C
6
to C
28
+ petroleum hydrocarbons in soil
as a series of aliphatic and aromatic carbon range fractions. The extraction method-
ology differs from other petroleum hydrocarbon methods because it uses n-pentane
and not methylene chloride as the extraction solvent. If methylene chloride is used
as the extraction solvent, aliphatic and aromatic compounds cannot be separated.
n-Pentane extracts petroleum hydrocarbons in this range efficiently. The whole
extract is separated into aliphatic and aromatic petroleum-derived fractions. This
group-type separation is based on SW-846 EPA Method 3611 (Alumina Column
Cleanup and Separation of Petroleum Wastes) and SW-846 EPA Method 3630 (Silica
Gel Cleanup). The aliphatic and aromatic fractions are analyzed separately by gas
chromatography, and quantified by summing the signals within a series of specified
carbon ranges that represent the fate and transport fractions. The gas chromato-
graph is equipped with a boiling point column (non-polar capillary column). GC
parameters allow the measurement of a hydrocarbon range of n-hexane (C
6
) to n-
octacosane (C
28
+), a boiling point range of approximately 65 °C to 450 °C.
The analytical method exploits the relationship between petroleum hydrocar-
bon fate and transport properties and boiling point discussed in TPHCWG Volume
3. The method has been specifically designed to resolve and quantify the 13
aliphatic and aromatic fate and transport fractions selected by the Working Group.
The Working Group also assigned toxicity criteria to each fraction by selecting tox-
icity data most representative of the fraction from the toxicology literature on
whole products, mixtures and individual petroleum constituents. When paired
with the Working Group toxicity criteria, the fate and transport fraction data can
be used to assess human health risk associated with exposures to petroleum-conta-
minated environmental media.
49

The method not only separates and quantifies aliphatic and aromatic fractions,
but it can also be used to obtain a TPH fingerprint for the sample. Experienced GC
analysts can use the fingerprint to identify the type of petroleum release, and the
extent of weathering. The method is currently written as a tiered approach with fin-
gerprint analysis preceding characterization of aliphatic and aromatic fractions. In
this way, the fingerprint can be used for a preliminary evaluation of the nature and
extent of the release.
The method is versatile and performance-based; therefore, it can be modified to
accommodate data quality objectives. It has been successfully applied to the char-
acterization of neat crude oil and petroleum products as well as soils containing
gasoline, JP-4, diesel and crude oil with different degrees of weathering.
Massachusetts EPH/VPH Approach
MA DEP published a regulatory framework for evaluating the TPH parameter in
human health risk assessments (MA DEP, 1994; Hutcheson, 1996). This framework
recommends using the EPH/VPH analytical procedure for petroleum hydrocarbon
mixtures. This procedure consists of two steps; quantification of volatile petroleum
hydrocarbons (VPH) and quantification of extractable petroleum hydrocarbons
(EPH). The VPH method includes the following analytes: benzene, toluene, ethyl-
benzene, and total xylenes (BTEX) as well as naphthalene and MTBE;
alkanes/cycloalkanes in the C
5
to C
8
and C
9
to C
12
carbon ranges; and
aromatics/alkenes in the C
9
to C
10
carbon range. The EPH method includes the fol-
lowing analytes: polycyclic aromatic hydrocarbons (PAHs); alkanes/cycloalkanes in
the C
9
to C
18
and C
19
to C
36
carbon range; and aromatics/alkenes in the C
10
to C
22
carbon range.
For the VPH fraction, the protocol for water samples involves direct purge and
trap and for soil samples involves a methanol extraction followed by purge and trap
concentration. The VPH methodology calls for a PID and FID in series. GC/PID is
recommended for separation and detection of the VPH target analytes and the C
9
to C
10
aromatic fraction, and the FID is used to detect the C
5
to C
8
and C
9
to C
12
aliphatic fractions. For the EPH fraction, the protocol for both water and soil
samples involves a methylene chloride extraction followed by Kuderna-Danish con-
centration. After solvent exchange to hexane, a Sep-pak (silica gel) cartridge and
two eluants (hexane followed by methylene chloride) are used to separate the
extract into the aliphatic and aromatic fractions. GC/FID is used as the detector
following concentration. Quantification of target analytes for both the VPH and
EPH procedures is done by comparing the area under the chromatogram from the
appropriate FID or PID response to the corresponding response of a standard
mixture containing the compounds of interest.
MA DEP developed the EPH/VPH methodology for specific application in
human health risk assessment. Similar to the Working Group’s fate and transport
fractions, MA DEP’s fractions have been assigned toxicity criteria based on refer-
ence compounds selected to represent each fraction. Cleanup standards have been
promulgated for the EPH/VPH fractions based on the toxicity of the reference
compounds, and the state has recommended the use of either or both EPH and
VPH analysis for a variety of petroleum mixtures.
50

APPENDIX I
Hydrocarbon Chemistry

HYDROCARBON CHEMISTRY
Petroleum and petroleum products are very complex mixtures that contain pri-
marily hydrocarbons (compounds containing molecules of carbon and hydrogen
atoms), heteroatom compounds (compounds containing molecules of carbon and
hydrogen atoms with heteroatoms such as sulfur, nitrogen, or oxygen), and rela-
tively small concentrations of metallic constituents. The complexity of petroleum
and petroleum products increases with carbon number. The heavier the material,
the larger the number of possible combinations of atoms. Gasoline has a smaller
number of components than diesel. For example, there are only 75 combinations
for molecules containing 10 carbons, but there are 366,319 possible combinations
for molecules containing 20 carbons. It is impossible to identify all components, so
petroleum and petroleum products are characterized in terms of boiling range and
approximate carbon number.
Regardless of the complexity, petroleum compounds can be generally classi-
fied into two major component categories: hydrocarbons and nonhydrocarbons.
Hydrocarbons comprise the majority of the components in most petroleum prod-
ucts and are the compounds that are primarily (but not always) measured as
TPH. The nonhydrocarbon components (those containing sulfur, nitrogen and
oxygen heteroatoms, as well as carbon and hydrogen in the molecule) are rela-
tively minor in most refined motor fuels as they tend to concentrate in the heavy
distillation fractions.
The hydrocarbon constituents can be grouped into saturated hydrocarbons, unsat-
urated hydrocarbons, and aromatics. There are several subclasses of importance
within these groups. Figure I-1 summarizes the different categories and subclasses.
SATURATED HYDROCARBONS
Saturated hydrocarbons are the primary class of compounds found in petroleum
and most petroleum products. They are comprised of single C-C bonds (with all
other remaining bonds saturated with H atoms.) The molecules can be arranged
in several configurations:
• Aliphatic: Straight or branched with the general formula:
C
n
H
2n+2
The common names for these types of compounds are alkanes and isoalka-
nes. The petroleum industry refers to these compounds as paraffins and
isoparaffins, respectively.
Examples:
CH
3
CH
2
CH
3
CH
3
CH
2
CH
2
CH
3
CH
3
Butane Isobutane
• Alicyclic: Cyclic compounds with the general formula:
C
n
H
2n
53
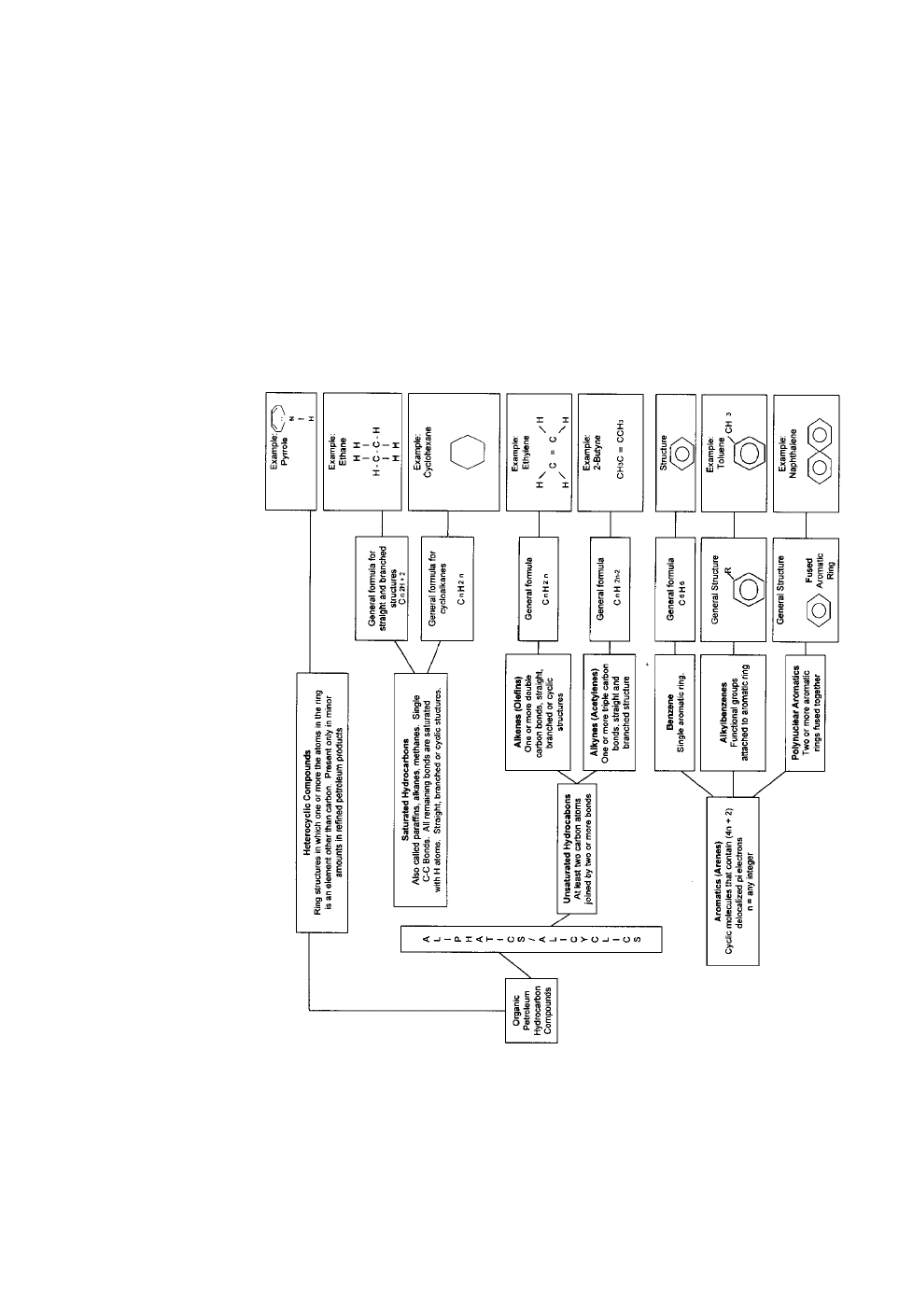
54
Figure I-1. TPH Compounds by Class.

These compounds are saturated hydrocarbons containing one or more rings
which may also contain saturated side chains. These compounds are also called
cycloalkanes. The petroleum industry commonly calls them naphthenes or
cycloparaffins.
Examples:
Cyclohexane Methylcyclopentane
UNSATURATED HYDROCARBONS
This class of compound has at least two carbon atoms in the molecule joined by two or
more bonds (C=C for alkenes, or C=
=
C for alkynes). These classes of compounds are
not found in crude oil and are produced primarily in cracking processes in the pro-
duction of smaller molecules from heavier ones. As molecules crack, they may form
double bonds when there is not enough hydrogen available to saturate the molecule.
Gasoline, for example may contain a significant amount of double-bonded carbon.
• Alkenes/Olefins: These compounds can be straight chain, branched, or
cyclic compounds. The general formula is:
C
n
H
2n
Examples:
H
2
C=CH
2
H
2
C=CHCH
2
CH
3
Ethylene 1-Butene
• Alkynes/Acetylenes: These compounds are found in straight chain and
branched structures. The general formula is:
C
n
H
2n-2
Examples:
HC=
=
CCH
3
HC =
=
CCH
2
CH
3
Acetylene 1-Butyne
AROMATICS
Aromatic compounds are a special class of unsaturated hydrocarbons. These com-
pounds are based on the benzene ring structure. The benzene ring contains six
carbons. Each carbon in the ring binds with one hydrogen, not typically shown in
structure diagrams. The benzene molecule can have one or more hydrogens sub-
stituted with side chains resulting in alkyl benzenes, or there may be two or more
aromatic rings fused together resulting in polycyclic aromatic hydrocarbons
(PAHs). All crudes and petroleum products (except some solvents produced from
petroleum) contain aromatics.
CH
3
55
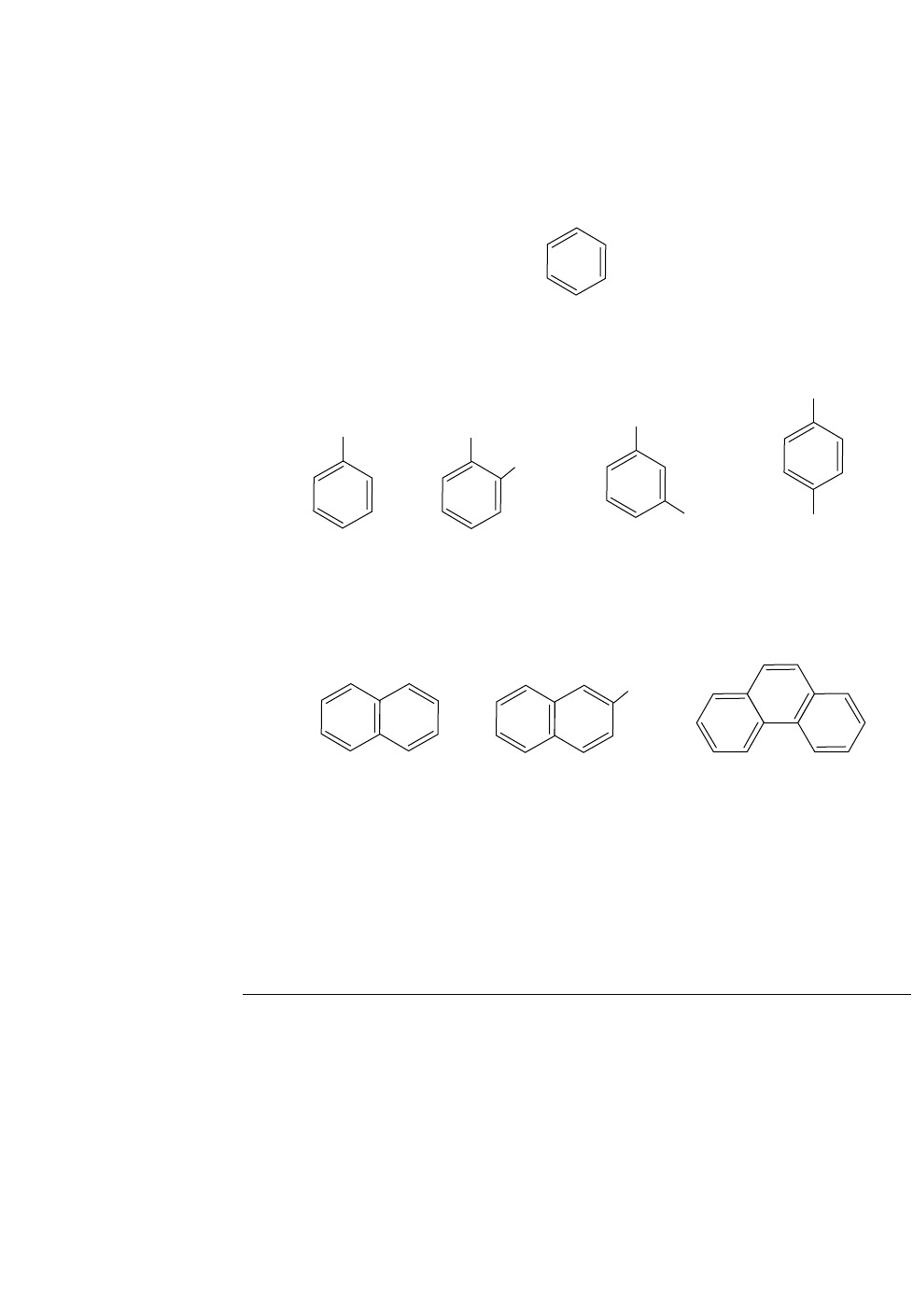
• Benzene: This is a single aromatic ring with the general formula:
C
n
H
n
Structure:
• Alkylbenzenes: These compounds have the base aromatic ring with side
chains attached.
Examples:
Toluene o-Xylene m-Xylene p-Xylene
• Polycyclic Aromatic Hydrocarbons (PAHs), are also known as polycyclic aro-
matic hydrocarbons. These compounds are two or more aromatic rings
fused together.
Examples:
Naphthalene 2-Methylnaphthalene Phenanthrene
Aromatics as a class are common environmental contaminants at petroleum
release sites. Monoaromatics, such as benzene, toluene, and xylenes have signifi-
cant water solubility, and are mobile in the environment. BTEX are volatile target
analytes in EPA methods. Several PAHs that can be found in petroleum and some
petroleum products can be persistent contaminants, particularly in soil and sedi-
ment matrices. PAHs are semivolatile target analytes in EPA methods.
NONHYDROCARBON COMPOUNDS
Crude oils contain significant amounts of organic nonhydrocarbon constituents,
mainly sulfur, nitrogen, and oxygen-containing compounds. There are also smaller
amounts of organometallic compounds and inorganic salts. These compounds are
concentrated in the heavier distillation fractions and residues during refining.
They are often referred to as asphaltenes.
Depending on the method used for determination of TPH, some of the sulfur,
nitrogen, and oxygen-containing compounds may be included in the TPH mea-
surements. By definition, these compounds are NOT hydrocarbons.
CH
3
CH
3
CH
3
CH
3
CH
3
CH
3
CH
3
CH
3
56
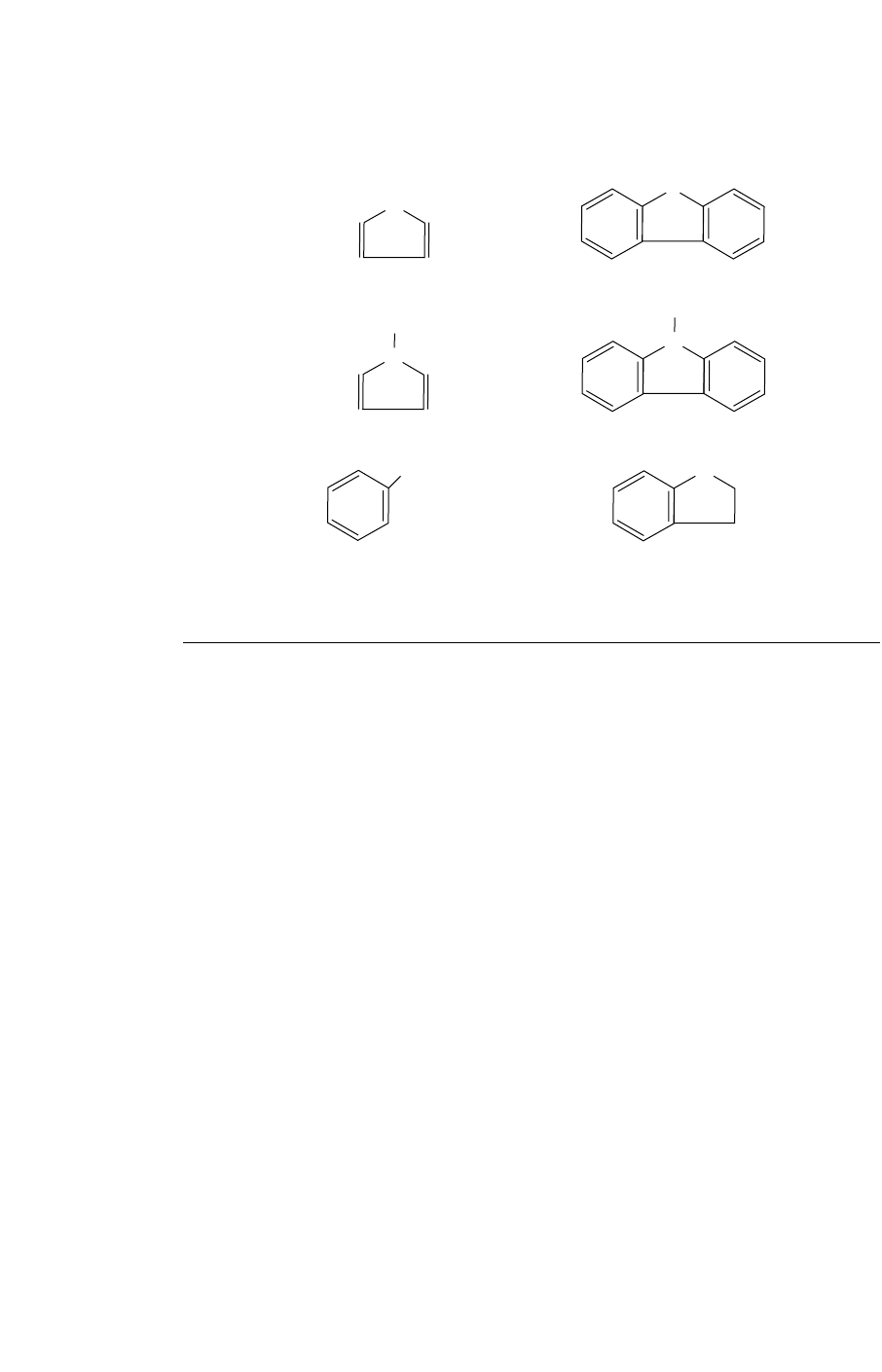
Examples:
Thiophene Dibenzothiophene
Pyrrole Carbazole
Benzoic Acid Benzofuran
SUMMARY
The composition of petroleum and petroleum products is extremely complex. This
complexity makes it difficult to determine environmental impacts at petroleum
release sites using traditional EPA methods. It should be very clear that a TPH mea-
surement cannot be used to evaluate risk. TPH methods measure a subset of the
sum of a complex mixture of compounds varying greatly in potential toxic prop-
erties. Target analyte methods measure only a few of the many compounds in
petroleum. Clearly, different analytical methods used for the determination of the
environmental impact of petroleum and petroleum products will provide very dif-
ferent information.
O
COOH
H
N
N
H
S
S
57

APPENDIX II
Characterization of Petroleum Products

APPENDIX II-A: PRODUCT COMPOSITION AND SPECIFICATION
A brief description of the most common petroleum products is included to assist
in understanding the composition of these materials and in the interpretation of
TPH for those cases in which the product type is known. These descriptions are
only general guidelines. Actual specifications for any given product may vary
depending on a variety of factors including specific refinery operations, market,
season, etc.
Petroleum products are either fractions from the distillation of crude oil or
blends of distillation fractions with reprocessed refinery streams. Complex refinery
operations govern the blending of different streams to produce the final commer-
cial products (See Appendix III for an illustration of how different products are
refined in a typical refinery).
Refinery operations are targeted to produce products that meet set specifica-
tions. A specification is a detailed listing of requirements and their acceptance
ranges. Specifications for petroleum products are performance driven. Tests
ensure the product has the appropriate physical and chemical properties for the
intended use. Specifications can change over time, based on regulatory pressures,
evolving manufacturing requirements, and sensitivity to environmental concerns.
Three major groups write specifications for products and materials: ASME
(American Society of Mechanical Engineers), ASTM (American Society for
Testing and Materials), and API (American Petroleum Institute). The American
Society for Testing and Materials has established specifications for most products
used on a large -scale basis in this country. It has sanctioned and approved numer-
ous standardized tests for measuring the properties established in petroleum
product specifications. Other specifications are written by manufacturers, end
users, and industry groups.
Petroleum products are used throughout our society from heavy military and
industrial end uses to large-scale uses in numerous consumer products.
Specifications required for the wide range of commercial petroleum products are
varied. Boiling range distribution, elemental composition, viscosity, flash point,
pour point, viscosity index, API gravity, specific gravity, color, ash content, water
content, demulsibility, as well as many other factors may be part of the specifica-
tions of a particular product for a specific use or application. While a physical prop-
erty such as boiling range may establish the initial product specification, other
finer specifications define their ultimate use in certain applications. The more spe-
cialized the application, the more detailed the list of specifications becomes for a
particular petroleum product.
GASOLINE
Automotive gasoline is a blended product. Refined to maximize cost-effectiveness,
it is manufactured from the C
4
to C
12
boiling range portions of distilled crude oil
and a variety of processed refinery streams. Depending on the source, a barrel of
crude oil can yield 10-40% gasoline from straight distillation. To increase yield and
performance, refinery processes crack large molecules and alkylate or polymerize
smaller molecules to produce molecules in the desired gasoline carbon range.
Some molecules that are in the right range but burn unevenly will be reformed or
61

isomerized to increase their octane value. Oxygenates that may be added to gaso-
line include methyl-tertiary butyl ether (MTBE), as well as alcohols. Gasoline con-
tains additives which may be added in relatively small amounts to enhance certain
performance characteristics.
The composition of gasoline is well characterized. It is a blended product of
many refinery streams. The potential number of isomers in this carbon range is rel-
atively small so that detailed analysis is possible. There are more than 200 individ-
ual components in the C
4
to C
12
boiling range. These hydrocarbons include 40 to
70% straight, branched and cyclic alkanes (normal paraffins, isoparaffins, and
naphthenes); alkenes (olefins) in variable concentrations but usually less than
10%; and 20 to 50% aromatics. The major components of gasoline that are targets
in EPA methods are benzene, toluene, ethylbenzene, xylenes, and naphthalene.
These compounds are sufficiently water soluble to leach to groundwater and are
thus mobile in the environment.
Gasoline can be produced to meet certain specifications. Its use demands that
the end product meet specifications for volatility, octane, boiling range distribu-
tion, sulfur content, gum content, water tolerance, corrosion resistance, and addi-
tive-derived properties. The American Society for Testing and Materials has a stan-
dard for motor gasolines, D 4814, which provides general guidelines for gasoline
quality. Volatility affects drivability and adequate starting. The volatility of a motor
gasoline must be such that enough low-boiling hydrocarbons are present in the
cold cylinders of the engine to form a flammable mixture. Octane indicates the
fuelís ability to prevent preignition or knocking in an engine. Octane ratings are
assigned to motor gasolines as an indication of the antiknock properties of the fuel.
Alkyl lead antiknock compounds, such as tetraethyl lead (TEL) and a manganese
tricarbonyl compound (MMT), were used as octane improvers until the advent of
unleaded gasolines in the mid 1970s when these additives were eventually phased
out. The proper octane levels are achieved in unleaded gasolines today by the use
of oxygenated additives (e.g., MTBE), higher concentrations of aromatic hydro-
carbons, and higher concentrations of branched chain hydrocarbons. These mol-
ecules have high research octane numbers (RON) and motor octane numbers
(MON). Octane numbers posted at gasoline dispensers are an average of the RON
and the MON. Additives blended into gasolines affect their overall performance
under many different conditions which exist during the operation of an automo-
bile or other vehicle. Some of the most commonly used gasoline additives (past
and present) are shown in Table II-A-1.
NAPHTHAS / SOLVENTS
Petroleum naphtha is a generic term for a variety of refined or unrefined petrole-
um products in the C
6
to C
12
range. Different naphthas can be all aromatic, all
paraffinic, aromatic/paraffinic combinations, or aromatic compounds such as
xylenes, etc. Another common term for naphtha is mineral spirits. A commonly
used type is Stoddard solvent that is typically in the C8 to C12 range. Naphthas are
widely used as diluents for paints, as solvents in dry-cleaning, for cutback asphalt,
and in extraction processes.
62

63
TABLE II-A-1. Common Gasoline Additives
Additive Composition Function
Oxidation inhibitors Aromatic amines and phenols Inhibit oxidation and gum formation
Corrosion inhibitors Carboxylic acids and carboxylates Inhibit corrosion of iron
Metal deactivators Chelating agent Inhibit oxidation and gum formation
catalyzed by certain metals, primarily
copper
Carburetor detergents Amines and amine carboxylates Prevent deposits in carburetor throttle body
Detergent-dispersants Polybutene succinimides Prevent deposits in carburetor throttle body
and intake manifold and ports
Deposit control additives Polybutene amines, polyether amines Remove and prevent deposits throughout
carburetor, intake manifold, and intake
ports and valves
Oxygenated blending agents Methyl-tert-butyl ether (MTBE), ethanol, Extend gasoline supply, increase octane
methanol, tert-butyl alcohol (TBA) number, environmental requirements,
reduce CO emissions
Antiicing additives Surfactants, alcohols, and glycols Prevent icing in carburetor and fuel system
Dyes Azo and other oil soluble compounds Identification
Antiknock compounds Lead alkyls, Organo-manganese compounds Increase octane number
Lead scavengers Ethylene dichloride (EDC), Reduce accumulation of inorganic lead
ethylene dibromide (EDB) deposits in the engine

Naphthas encompass a wide variety of volatile hydrocarbons with varying physi-
cal properties. The initial boiling point may be as low as 80°F; and end points may
reach 450°F, depending on the application. A more narrow boiling range is speci-
fied by ASTM for aliphatic and aromatic naphthas. According to ASTM standards
for naphthas in D 3734 and D 3735, there are five basic types of naphthas. Three
are classified as VM&P naphthas that are primarily aliphatic in composition and
classified as Type I-regular (minimum flash point of 40°F); Type II-high flash
(minimum flash point of 80°F; and Type III-odorless (minimum flash point of
40°F). There are two types of high-flash aromatic naphthas: Type I, which is known
as Aromatic 100, has a minimum flash point of 100°F; and Type II, which is known
as Aromatic 150, has a minimum flash point of 150°F.
Specifications for naphthas include color, odor, sulfur content, aromaticity, flash
point, specific gravity, initial boiling point, Kauri-butanol value, copper corrosion,
bromine number, and appearance.
AVIATION GASOLINE
Aviation gasolines have strictly limited hydrocarbon compositions with the highest
octane economically possible within a specific boiling range. Aviation gasolines are
comprised of 50-60% saturated hydrocarbons (paraffins and isoparaffins), 20-30%
cycloalkanes (naphthenes), approximately 10% aromatics, and usually no olefins.
Most aviation gasolines are made by blending selected straight-run naphtha frac-
tions with isopentane and alkylate. All aviation gasolines are leaded and use
tetraethyllead (TEL) with ethylene dibromide (EDB). Table II-A-2 lists additives
typically used in aviation gasoline.
There are different grades of aviation gasolines that can be distinguished by the
color dye additives and the lead content. Examples of designations of aviation gaso-
line include Grade 80 (red), Grade 100 (green), and Grade 100LL (blue).
JET FUELS
There are two general grades of jet fuel:
• a wide-cut heavy naphtha-kerosene blend previously used by the U.S. Air
Force as JP-4; and
• a kerosene used by the world’s airlines as Jet A or Jet A-1, by the U.S. Navy as JP-
5, and currently by the U.S. Air Force as JP-8. JP-5 is a more narrow cut of JP-8.
JP-4 is a type of aviation fuel that is currently being phased out of use in military
aircraft. It is a mixture of naphtha, gasoline, and kerosene hydrocarbons. The
typical carbon range includes C
6
to C
14-16
.
Jet A fuels are essentially a fraction distilled from crude oil mixed with some
cracked material. These products contain saturated hydrocarbons (80 to 90%),
aromatics (10 to 20% for jet fuel and as high as 30% for kerosene), and do not gen-
erally contain alkenes (olefins). Sulfur compounds are removed and any alkenes
are saturated by hydrotreating. Jet fuels (Jet A), like kerosenes, are primarily com-
prised of hydrocarbons in the C
8
to C
17
range with the majority in the C
10
to C
14
range. There is no detailed analysis of the components in this range because the
64

65
TABLE II-A-2. Aircraft Fuel Additives
Additive Composition Function
Aviation Gasoline
Antiknock Lead alkyls Increase octane number
Oxidation Inhibitor Alkylated amines aromatic phenols Inhibit oxidation
Dyes Azo and other oil soluble compounds Identification
Lead scavenger Ethylene Dibromide Reduce accumulation of inorganic led deposits
in the engine
Jet Fuel
Antirust Alkyl amine salts of orthophosphoric acid, Minimize formation of rust
polyoxyethylated compounds, diamines
and amides
Antiicing Ethylene glycol monomethyl ether, Prevent ice formation
glycol ethers
Antioxidation Alkyl phenols, arylamines Provide oxidation stability
Antistatic Pure paraffinic hydrocarbons, Shell’s Decrease specific resistivity of the fuel to lesson
ASA addative (Chromium salt of alkylated the incidence of fires
salicylic acid, calcium disulfosucccinimide
and vinyl methacrylate copolymer)
Biocide Organoboron compounds Inhibit hte growth of bacteria and fungi in
storage tanks
Source: McGraw-Hill Encyclopedia of Science and Technology and Corrosion Inhibitors, an offical NACE Publication, 1973.

product is processed to meet boiling range and performance characteristic speci-
fications. The number of isomers in this carbon range precludes detailed identifi-
cation of individual components.
All jet fuels meet stringent specifications necessary for the performance require-
ments of aircraft turbine engines and fuel systems. These fuels must meet specifi-
cations of extreme cleanliness, freedom from oxidation in high-temperature zones,
atomization and ignition at low temperatures, fluidity at low temperatures, com-
bustion quality, smokeless and burn with adequate heat release. In addition, jet
fuel must be dry and have an extremely low freezing point. It must also contain an
electrical conductivity additive to ensure rapid dissipation of charge, which can
cause ignition of the product resulting in fires.
KEROSENE
Kerosene is essentially the same boiling range distillation fraction as Jet A and thus
has the same hydrocarbon composition. There are some differences in specifica-
tions. Most commercial kerosenes are sold under two general classes: No. 1-K and
No. 2-K. No. 1-K kerosene is a low-sulfur grade kerosene while No. 2-K is a regular
grade of kerosene with a higher sulfur content. Additional specifications for
kerosene include low aromatics content (except in the case of tractor fuel), low vis-
cosity, a flash point greater than 73°F, and a melting point no more than -25°F.
Kerosene with high paraffin content is used in oil lamps.
DIESEL FUEL
Transportation diesels are manufactured primarily from distilled fractions of crude
oil with some blending with cracked gas oils. The major components of diesels are
similar to those present in the crude oil, but include a higher fraction of aromatics
(up to 30 to 40%). Diesel fuel is essentially the same as furnace oil, but the propor-
tion of cracked gas oil is usually less than in furnace oil. Although cracking process-
es also produce small alkenes as well as aromatics, the small alkenes are not in the
diesel carbon range and end up in the gasoline pool. The typical carbon range for
diesel #1 grades is C
8
to C
17
range, with the majority in the C
10
to C
14
range (similar
to Jet A and kerosene). The typical carbon range for diesel # 2 fuels is C
8
to C
26
,
with the majority in the C
10
to C
20
range (similar to fuel oil No. 2). In all cases, the
majority of the fuels is 60-90% normal, branched, and cyclic alkanes.
These middle petroleum distillates are classified primarily by the ASTM for the
intended primary applications. The specifications are usually for a defined volatil-
ity, boiling range, and sulfur content. There are five different grades of diesel fuel
oils for uses that range from automobiles, commercial trucks, buses, to marine and
railroad engines. Additional specifications include requirements for viscosity, ash
content, copper strip corrosion rating, cetane number (a measure of the ignition
quality that influences starting and combustion roughness, analogous to octane
number for gasoline), cetane index, cloud point, aromaticity, water and sediment
content, and carbon residue. Additives are used in diesel fuels to protect the fuel
system against deposits, rust and corrosion, to keep the fuel system components
clean, and to improve diesel fuel cetane (Table II-A-3). Many of these specifications
are outlined in ASTM D 975.
66

67
TABLE II-A-3. Typical Classes of Diesel Fuel Additives
Additive Composition Function
Dispersant Succinate esters, succinimides, Mannich bases Extend filter life, disperse particulates and
sediment
Detergent Synthetic sulfonates, phenates, salicylates, Prevent deposits
phosphonates
Antirust Ethoxylated alkyl phenols, alkenyl succinic Prevent rust
acids, amine phosphates
Cetane Number Improver Alkyl nitrates, hydroperoxides Improve burning characteristics of fuel
Biocide Organoboron compounds Inhibit the proliferation of bacteria and fungi
Pour Point Depressant Polymethacrylates, alkylated naphthaenes, Reduce the yield stress of the fuel and
ethylene vinyl acetate copolymers, fumarate- improve pumpability at low temperatures
vinylacetate copolymers, alkylated polystyrene,
acylated polystyrene, polyolefins,aliphatic
amine oxides,and oxidized wax
Source: McGraw Hill Encyclopedia of Science and Technology.

FUEL OILS
Fractions from crude oil distillation that are heavier than diesel/middle distillates
are often called residual fuel oils. Other commonly used names are No. 4, 5, and 6
fuel oils and bunker C. These oils can be used as fuel or as feed to refinery conver-
sion units to produce lighter, more valuable fuel fractions. The sources of fuel oils
may be directly from the distillation process and/or a complex process of selection
and blending of various petroleum fractions to meet definite specifications. In prin-
ciple, these materials start in the C
20
to C
25
range and can go higher than C
40
.
Blending with lower molecular weight fractions to decrease viscosity of heavier frac-
tions may widen the carbon range to as low as C
6
. The composition of these oils is
quite variable but they all contain saturated and aromatic hydrocarbons (including
PAHs) as well as nonhydrocarbons (heteroatom containing molecules).
There are seven different grades of fuel oil which are primarily used in different
types of fuel burning equipment under different conditions of operation. They are
grades 1& 2, grades 4 (light) and 4, grades 5 (light) and 5 (heavy), and 6. The specifi-
cations governing fuel oils are similar to those of diesel fuels with some variations.
Maximum and minimum specifications for flash point, viscosity, API gravity, ash
content, water and sediment contents, distillation end point and amount recovered,
sulfur content, cetane number, copper strip corrosion rating, aromaticity, cloud point,
and carbon residue are outlined in the standard ASTM D 396 specification for fuel oils.
LUBRICATING OILS
Lubricating oils have very high boiling points (>650°F). Typical carbon ranges are
C
20
to C
45
+. Normal paraffins (straight chain alkanes) are usually removed by
solvent extraction. These materials are enriched in cycloparaffins, aromatics, and
nonhydrocarbons and are best characterized on the basis of physical properties
such as refractive index, density, and molecular weight. They may contain 70 to
90% alkanes and 10 to 30% aromatics. Greases are lubricating oils to which a thick-
ening agent has been added. Soaps are common additives. See Table II-A-4.
Lubricating oils encompass a wide variety of commercial products used in
numerous applications. The carbon number range of a lubricating oil determines
the type of application. Refinery processes as well as the type of crude feed deter-
mine the type and quality of lubricating oil base stocks that can be used for various
applications. Lubricating oils run the gamut of products from automotive and avi-
ation oils, railroad lubricants, marine lubricants, industrial oils including turbine
oils, hydraulic oils, cylinder oils, compressor oils, refrigeration oils, and many
others too numerous to include here.
The following test specifications may be required for particular lubricating oil for-
mulations: Aniline point, ash and sulfated ash, color, ASTM D 1500, copper strip
corrosion, demulsibility, dielectric strength, flash point, fire point, API gravity, inter-
facial tension, load-carrying ability, neutralization number, oxidation stability, pour
point, cloud point, viscosity, and viscosity index. All of these tests help govern the
quality of a lubricating oil for a particular application. In some cases, the applica-
tion dictates the quality of the base stocks utilized, since functionality is a by-product
of base stock quality. ASTM D 4485 gives a summary of the specifications for the per-
formance of engine oils. Similar standards for lubricating oils are set by manufac-
turers of machinery requiring lubricants that conform to particular specifications.
68
69
TABLE II-A-4. Lubricant Additive Utilization
Lubricant Type
Metal
Additive Engine Transmission Axle Hydraulic Gear Turbine Working
Type Oil Fluids Oils Oils Oils Oils Fluids
Metallic detergents •
Ashless disperants • •
Anti-oxidants •••••••
Anti-wear agents •••••
Rust inhibitors • • • •
Corrosion inhibitors • • • •
Friction modifiers • • • •
Extreme pressure agents • • •
Anti-foam agents •••••••
Viscosity improvers • •
Pour point depressants • • •
Seal swell • •

SUMMARY OF PETROLEUM PRODUCTS COMPOSITION AND SPECIFICATIONS
The composition of gasoline range materials is well characterized. Beyond the
gasoline range, the number of isomers is so great that it is not possible to deter-
mine individual compounds. In addition, the production of these materials does
not require knowledge of detailed compositional information since they are per-
formance-based defined products.
APPENDIX II-B ENVIRONMENTAL FATE OF PETROLEUM
PRODUCTS: WEATHERING AND TRANSPORT
Petroleum products released into the environment undergo weathering processes
with time. These processes include evaporation, leaching (transfer to the aqueous
phase) through solution and entrainment (physical transport along with the
aqueous phase), chemical oxidation, and microbial degradation (Christensen and
Larsen, 1993). The rate of weathering is highly dependent on environmental con-
ditions. For example, gasoline, a volatile product, will evaporate readily in a surface
spill, while gasoline released below 10 feet of clay topped with asphalt will tend to
evaporate slowly (weathering processes may not be detectable for years).
Evaporative processes are very important in the weathering of volatile petroleum
products, and may be the dominant weathering process for gasoline. Automotive
gasoline, aviation gasoline, and JP-4 contain 20% to 99% highly volatile (less than
9 carbon atom) components. Figure II-B-1 shows four gas chromatograms of gaso-
lines that have undergone different amounts of evaporation. The four GC/FID
chromatograms in Figure II-B-1 show a dramatic change in composition from the
unweathered sample (0% evaporated) to the highly weathered sample (98% evap-
orated). In each chromatogram, the peaks from 0 to 2 minutes are from a dilution
solvent. The most volatile gasoline components elute from (or pass through) the
GC between two and seven minutes (visible in the very front of a chromatogram).
The GC/FID traces show the loss of these components as the gasoline evaporates.
Leaching processes introduce hydrocarbon into the water phase by solubility
and entrainment. Aromatics, and especially BTEX, tend to be the most water-
soluble fraction of petroleum (Senn and Johnson, 1987). Petroleum contaminated
groundwater tends to be enriched in aromatics relative to other petroleum con-
stituents. Relatively insoluble hydrocarbons may be entrained in water through
adsorption into kaolinate particles suspended in the water or as an agglomeration
of oil droplets (microemulsion) (Coleman et al., 1984). In cases where groundwa-
ter contains only dissolved hydrocarbons, it may not be possible to identify the orig-
inal petroleum product because only a portion of the free product will be present
in the dissolved phase. As whole product floats on groundwater, the free product
will gradually lose the water-soluble compounds. Whole products have highly dis-
tinctive GC fingerprints relative to water-soluble fractions. Groundwater containing
entrained product will have a GC fingerprint that is a combination of the free
product chromatogram plus enhanced amounts of the soluble aromatics.
Leaching processes of petroleum products in soils can have a variety of potential
scenarios. Part of the aromatic fraction of a petroleum spill in soil may partition
into water that has been in contact with the contamination. Evaporative processes,
70
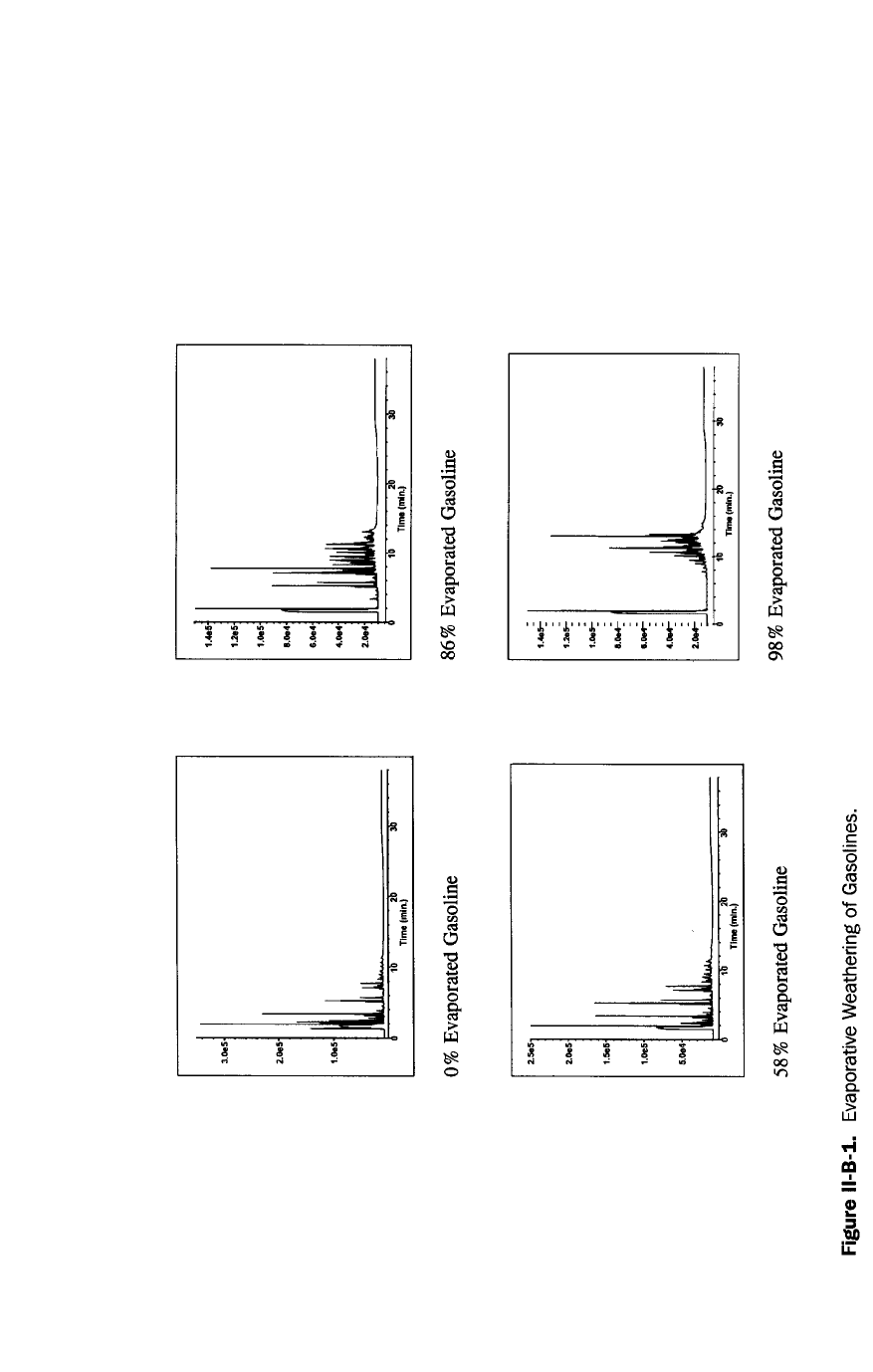
71
Figure II-B-1. Evaporative Weathering of Gasolines.
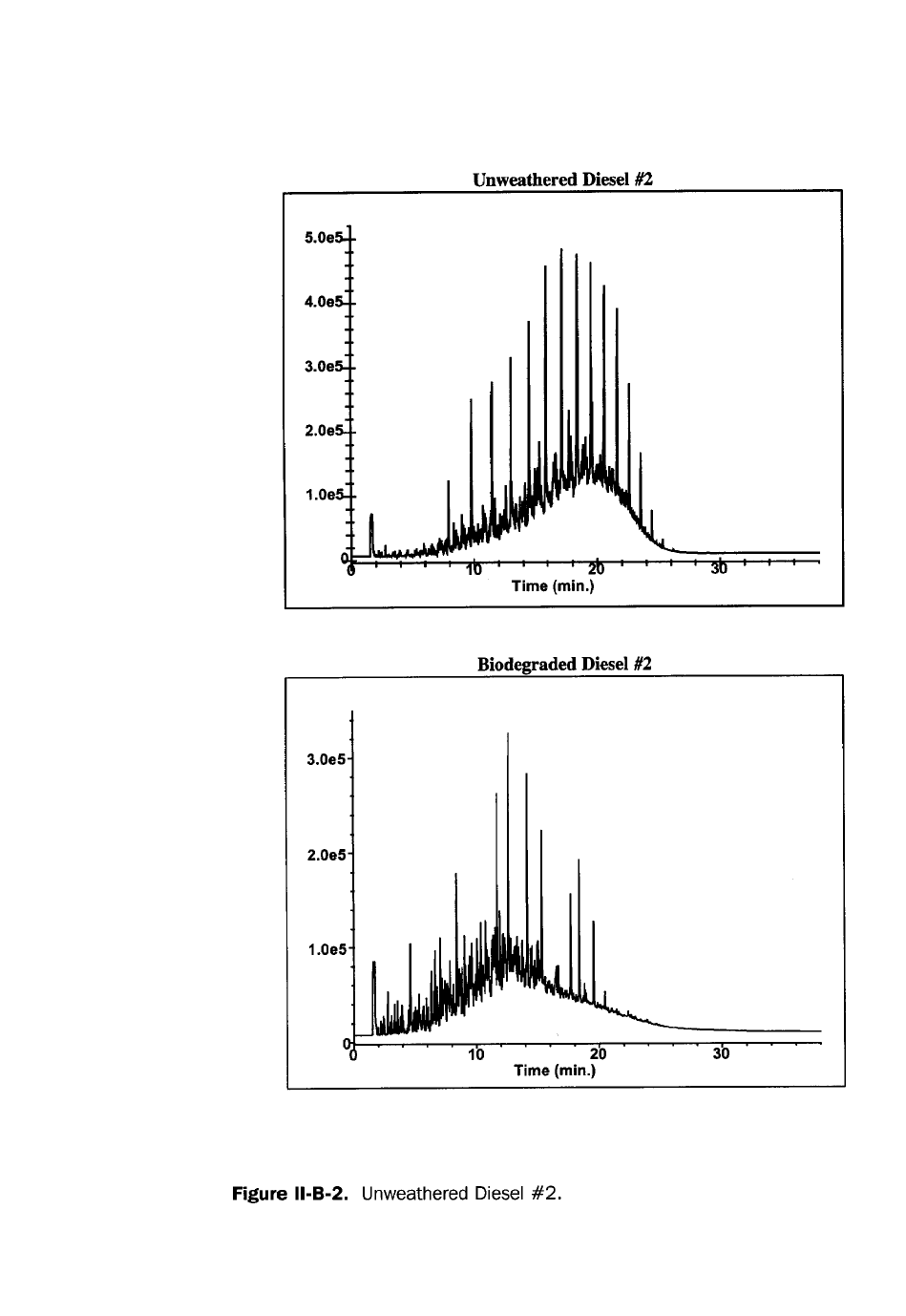
72
Figure II-B-2. Unweathered Diesel #2.

however, are also working simultaneously to remove the volatile aromatics such as
benzene and toluene. A GC characterization of a wet soil sample may sometimes
show an unusually high aromatic content.
It is not unusual for a plume of water-soluble aromatics to radiate out from the
origin of a spill. Storm drains, pipes, and utility lines may provide conduits for
water and water soluble hydrocarbons. Generally, entrained hydrocarbon is found
in water close to the petroleum source, while dissolved aromatics may be found
quite far from the origin of a spill. Oxygenates, such as methyl-t-butyl ether
(MTBE), are even more water soluble than aromatics and are highly mobile in the
environment.
Biodegradation processes can be very complex. The extent of biodegradation is
dependent on many factors including the type of microorganisms present, envi-
ronmental conditions (temperature, oxygen levels, moisture etc.), predominant
hydrocarbon types, and bioavailability of hydrocarbon contaminants. Evidence
indicates that the primary factor controlling the extent of biodegradation is the
molecular composition of the soil contaminant. Multiple ring cycloalkanes are
hard to degrade, while PAHs display varying degrees of degradation. n-Alkanes
biodegrade rapidly with branched alkanes and single saturated ring compounds
degrading more slowly. Wang et al. (1994) found that samples from a 22-year-old
spill of Bunker C oil were >80% depleted in PAHs and that the naphthalenes
(diaromatics) were heavily depleted relative to the PAHs.
Figure II-B-2 shows one GC/FID chromatogram of fresh diesel #2 and a second
GC/FID chromatogram of a weathered diesel #2. The unweathered diesel #2 shows
a distinctive pattern of n-alkanes. The weathered diesel is depleted of n-alkanes, but
displays a characteristic pattern of branched alkanes. Signals of two of the
branched alkanes, pristane (tetramethyltridecane) and phytane (tetramethylte-
tradecane), are sometimes compared to signals of their neighboring C
17
and C
18
n-
paraffins, respectively, to evaluate the progress of a bioremediation effort.
An understanding of weathering processes is valuable to environmental test lab-
oratories, bioremediation engineers, and risk assessors. Weathering changes
product composition and may affect testing results, the ability to bioremediate, and
the toxicity of the spilled product. Unfortunately, the database available on the
composition of weathered products is limited.
73

APPENDIX III
Refinery Flow Diagrams
76 77
78 79

APPENDIX IV
Quick Reference of TPH Methods
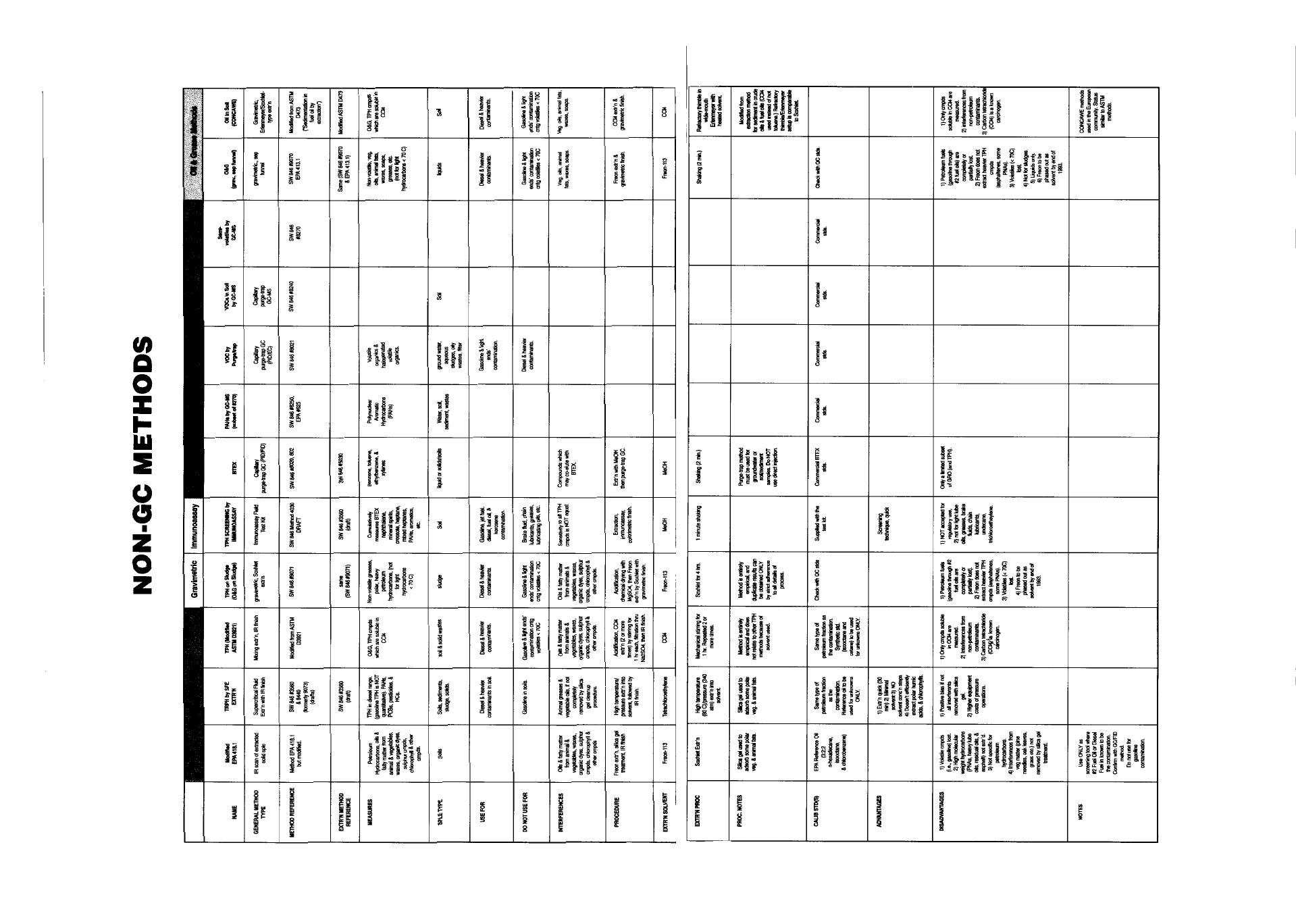
82 83
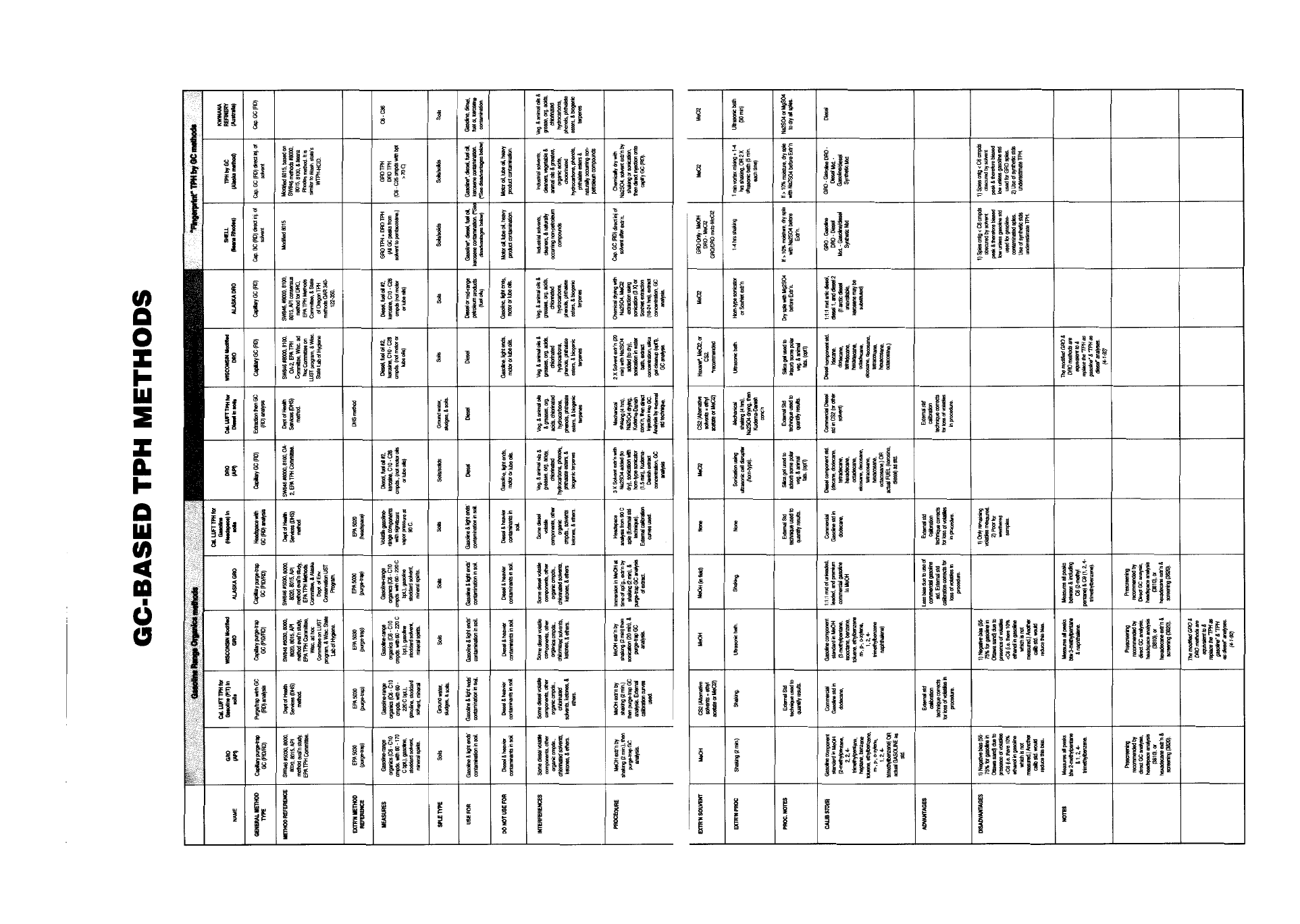
84 85

EXPLANATION OF ACRONYMS
AAR American Association of Railroads
AEHS Association for the Environmental Health of Soils
API American Petroleum Institute
ASME American Society of Mechanical Engineers
ASTM American Society for Testing and Materials
BP British Petroleum
BTEX Benzene, Toluene, Ethylbenzene, and Xylenes
DOD Department of Defense
DOE Department of Energy
DOT Department of Transportation
DRO Diesel Range Organics
EDB Ethylene Dibromide
EDC Ethylene Dichloride
EPH Extractable Petroleum Hydrocarbons
FID Flame Ionization Detector
GC Gas Chromatography
GC/MS Gas Chromatography/Mass Spectrometry
GRI Gas Research Institute
GRO Gasoline Range Organics
HPLC High Performance Liquid Chromatography
IR Infrared
LC Liquid Chromatography
MMT Methylcyclopentadienyl Manganese Tricarbonyl
MON Motor Octane Number
MTBE Methyl Tertiary-butyl Ether
NACE National Association of Corrosion Engineers
NPDES National Pollutant Discharge Elimination System
O&G Oil and Grease
PAH Polycyclic Aromatic Hydrocarbon
PHC Petroleum Hydrocarbon
PIANO Paraffins, Isoparaffins, Naphthenes, Olefins, and Aromatics
PID Photoionization Detector
87

PNA Polynuclear Aromatic
PPM Part Per Million
RBCA Risk-Based Corrective Action
RON Research Octane Number
SFE Supercritical Fluid Extraction
SPE Solid Phase Extraction
SPME Solid Phase Micro-extraction
SVOC Semi-volatile Organic Compound
TBA Tert-butyl Alcohol
TEL Tetraethyl Lead
TLC Thin Layer Chromatography
TPH Total Petroleum Hydrocarbons
TPHCWG Total Petroleum Hydrocarbon Criteria Working Group
TPH-D Total Petroleum Hydrocarbons-Diesel
TPH-G Total Petroleum Hydrocarbons-Gasoline
TPHV Total Petroleum Hydrocarbons Volatiles
TRPH Total Recoverable Petroleum Hydrocarbons
UCM Unresolved Complex Mixture
USEPA United States Environmental Protection Agency
UV Ultraviolet
VM&P Varnish Maker’s and Painter’s [naptha], i.e. paint thinner
VOC Volatile Organic Compound
VPH Volatile Petroleum Hydrocarbons
88

GLOSSARY OF TERMS
Additive A substance added to petroleum mixtures (e.g., lubricating
oils) to impart new or to improve existing characteristics.
Aliphatic hydrocarbon Hydrocarbons in which the carbon-hydrogen groupings
are arranged in open chains which may be branched. The
term includes paraffins and olefins and provides a distinc-
tion from aromatics and naphthenes, which have at least some
of their carbon atoms arranged in closed chains or rings.
Alkanes Hydrocarbons that contain only single bonds. The chem-
ical name indicates the number of carbon atoms and ends
with the suffix “–ane”.
Alkenes Hydrocarbons that contain carbon-carbon double bonds.
The chemical name indicates the number of carbon
atoms and ends with the suffix “ene”.
Alkyl groups A group of carbon and hydrogen atoms that branch from
the main carbon chain or ring in a hydrocarbon mole-
cule. The simplest alkyl group, a methyl group, is a carbon
atom attached to three hydrogen atoms.
Analyte The chemical for which a sample is tested, or analyzed.
Antibody A molecule having chemically reactive sites specific for
certain other molecules.
Aromatic A compound containing one or more conjugated rings
that also may contain sulfur, nitrogen, and oxygen.
Asphaltene A constituent of petroleum products with a high molecu-
lar mass and dark color, insoluble in n-heptane, and
soluble in hot benzene.
ASTM American Society for Testing and Materials, responsi-
ble for many of the standard methods used in the
petroleum industry.
Biogenic Bacterial or vegetation derived material.
Biological lipids Any biological fluids that are miscible with a nonpolar
solvent. These materials include waxes, essential oils,
chlorophyll, etc.
Boiling point A characteristic physical property of a liquid at which the
vapor pressure is equal to that of the atmosphere and the
liquid is converted to a gas.
89

BTEX Benzene, toluene, ethylbenzene, and the xylene isomers.
Bunker fuel Heavy residual oil, also called bunker C, bunker C fuel oil,
or bunker oil.
Chromatogram The resultant electrical output of sample components
passing through a detection system following chromato-
graphic separation. A chromatogram may also be called a
trace.
Cleanup A preparatory step following extraction of a sample media
designed to remove components that may interfere with
subsequent analytical measurements.
Confirmation column A secondary column in chromatography that contains a
stationary phase having different affinities for compo-
nents in a mixture than in the primary column. Used to
confirm analyses that may not be completely resolved
using the primary column.
Cracking A process whereby the relative proportion of lighter or
more volatile components of an oil is increased by changing
the chemical structure of the constituent hydrocarbons.
Crude oil Naturally occurring mixture consisting essentially of many
types of hydrocarbons, but also containing sulfur, nitro-
gen or oxygen derivatives. Crude oil may be of paraffinic,
asphaltic or mixed base, depending on the presence of
paraffin wax and bitumen in the residue after atmospheric
distillation. Crude oil composition varies according to the
geological strata of its origin.
Cut The distillate obtained between two given temperatures
during a distillation process.
Cycloalkane A class of alkanes that are in the form of a ring.
Cycloparaffin An example of a cycloalkane.
Diesel fuel That portion of crude oil that distills out within the tem-
perature range approximately 200-370°C. A general term
covering oils used as fuel in diesel and other compression
ignition engines.
Distillate A product obtained by condensing the vapors evolved
when a liquid is boiled and collecting the condensation in
a receiver that is separate from the boiling vessel.
Distillation range A single pure substance has one definite boiling point at
a given pressure. A mixture of substances, however,
exhibits a range of temperatures over which boiling or
distillation commences, proceeds and finishes. This range
of temperatures, determined by means of standard appa-
ratus, is termed the ‘distillation’ or ‘boiling’ range.
90

Eluate The solutes, or analytes, moved through a chromato-
graphic column (see elution).
Eluent Solvent used to elute sample.
Elution Process whereby a solute is moved through a chromato-
graphic column by a solvent (liquid or gas), or eluent.
Extract In solvent extraction, the portion of a sample preferen-
tially dissolved by the solvent and recovered by physically
separating the solvent.
“Fingerprint” analysis A direct injection GC/FID analysis in which the detector
output - the chromatogram - is compared to chro-
matograms of reference materials as an aid to product
identification.
Flame ionization A detector for a gas chromatograph that measures any
detector (FID) thing that can burn.
Fuel oil A general term applied to oil used for the production of
power or heat. In a more restricted sense, it is applied to
any petroleum product that is used as boiler fuel or in
industrial furnaces. These oils are normally residues, but
blends of distillates and residues are also used as fuel oil.
The wider term, ‘liquid fuel,’ is sometimes used, but the
term ‘fuel oil’ is preferred.
Gas chromatography An analytical technique, employing a gaseous mobile
phase, that separates mixtures into their individual com-
ponents.
Gas oil A petroleum distillate with a viscosity and distillation range
intermediate between those of kerosene and light lubricating oil.
Gasoline (petrol) Refined petroleum distillate, normally boiling within the
limits of 30-220°C, which, combined with certain addi-
tives, is used as fuel for spark-ignition engines. By exten-
sion, the term is also applied to other products that boil
within this range.
Gravimetric Gravimetric methods weigh a residue.
Grease A semisolid or solid lubricant consisting of a stabilized
mixture of mineral, fatty, or synthetic oil with soaps, metal
salts, or other thickeners.
Headspace The vapor space above a sample into which volatile mole-
cules evaporate. Certain methods sample this vapor.
Heating oil Gas oil or fuel oil used for firing the boilers of central
heating systems.
91

Hydraulic fluid A fluid supplied for use in hydraulic systems. Low viscosity
and low pour-point are desirable characteristics. Hydraulic
fluids may be of petroleum or nonpetroleum origin.
Hydrocarbons Molecules that consist only of hydrogen and carbon atoms.
Immunoassay Portable tests that take advantage of an interaction between
an antibody and a specific analyte. Immunoassay tests are
semi-quantitative and usually rely on color changes of
varying intensities to indicate relative concentrations.
Infrared spectroscopy An analytical technique that quantifies the vibration
(stretching and bending) that occurs when a molecule
absorbs (heat) energy in the infrared region of the elec-
tromagnetic spectrum.
Jet fuel Kerosene or gasoline/kerosene mixture for fueling aircraft gas
turbine engines.
Kerosene A refined petroleum distillate intermediate in volatility
between gasoline and gas oil. Its distillation range general-
ly falls within the limits of 150 and 300°C. Its main uses
are as a jet engine fuel, an illuminant, for heating pur-
poses, and as a fuel for certain types of internal combus-
tion engines.
Light distillate A term lacking precise meaning, but commonly applied
to distillates, the final boiling-point of which does not exceed
300°C.
Liquid chromatography A chromatographic technique which employs a liquid
mobile phase.
Liquid/liquid extraction An extraction technique in which one liquid is shaken
with or contacted by an extraction solvent to transfer mol-
ecules of interest into the solvent phase.
Mass spectrometer An analytical technique that “fractures” organic com-
pounds into characteristic “fragments” based on func-
tional groups that have a specific mass-to-charge ratio.
Middle distillate One of the distillates obtained between kerosene and lubri-
cating oil fractions in the refining processes. These include
light fuel oils and diesel fuels.
Mineral hydrocarbons Petroleum hydrocarbons, considered “mineral” because they
come from the earth rather than from plants or animals.
Mobile phase In chromatography, the phase (gaseous or liquid) respon-
sible for moving an introduced sample through a porous
medium to separate components of interest.
92

Naphtha Straight-run gasoline fractions boiling below kerosene and
frequently used as a feedstock for reforming processes. Also
known as heavy benzine or heavy gasoline.
Naphthene Petroleum industry term for a cycloparaffin (cycloalkane).
Olefin Synonymous with alkene.
Oxygenated gasolines Gasolines with added ethers or alcohols, formulated
according to the Federal Clean Air Act to reduce carbon
monoxide emissions during winter months.
Polycyclic aromatic PAHs consist of a suite of compounds comprised of two or
hydrocarbons (PAHs) more aromatic rings. PAHs are found in many petroleum
mixtures, and they are predominantly introduced to the
environment through natural and anthropogenic com-
bustion processes.
Paraffin (alkanes) One of a series of saturated aliphatic hydrocarbons, the
lowest numbers of which are methane, ethane, and
propane. The higher homologues are solid waxes.
Partitioning In chromatography, the physical act of a solute having dif-
ferent affinities for the stationary and mobile phases.
Partition ratios, K, are defined as the ratio of total analyt-
ical concentration of a solute in the stationary phase, C
S
,
to its concentration in the mobile phase, C
M
.
Photoionization A gas chromatographic detection system which utilizes an
detector (PID) ultraviolet lamp as an ionization source for analyte detec-
tion. It is usually used as a selective detector by changing
the photon energy of the ionization source.
Positive bias A result that is incorrect and too high.
Purge and trap A chromatographic sample introduction technique in
volatile components which are “purged” from a liquid
medium by bubbling gas through it. The components are
then concentrated by “trapping” them on a short interme-
diate column, which is subsequently heated to drive the
components on to the analytical column for separation.
Purge gas Typically helium or nitrogen, used to remove analytes
from the sample matrix in purge/trap extractions.
Retention time The time it takes for an eluate to move through a chro-
matographic system and reach the detector. Retention
times are reproducible and can therefore be compared to
a standard for analyte identification.
Separatory funnel Glassware shaped like a funnel with a stoppered rounded
top and a valve at the tapered bottom, used for
liquid/liquid separations.
93

Soap An emulsifying agent made from sodium or potassium
salts of fatty acids.
Solvent Fluids in which certain kinds of molecules dissolve. While
they typically are liquids with low boiling points, they may
include high-boiling liquids, supercritical fluids, or gases.
Sonication A physical technique employing ultrasound to intensely
vibrate a sample media in extracting solvent and to maxi-
mize solvent/analyte interactions.
Soxhlet extraction An extraction technique for solids in which the sample is
repeatedly contacted with solvent over several hours,
increasing extraction efficiency.
Stationary phase In chromatography, the porous solid or liquid phase
through which an introduced sample passes. The different
affinities the stationary phase has for a sample allow the
components in the sample to be separated, or resolved.
Supercritical fluid An extraction method where the extraction fluid, usually
extraction CO
2
, is present at a pressure and temperature above its
critical point.
Target analyte Target analytes are compounds that are required analytes
in U.S. EPA analytical methods. BTEX and PAHs are
examples of petroleum-related compounds that are target
analytes in U.S. EPA Methods.
Thin layer chroma- A chromatographic technique employing a porous
tographiy (TLC) medium of glass coated with a stationary phase. An
extract is spotted near the bottom of the medium and
placed in a chamber with solvent (mobile phase). The
solvent moves up the medium and separates the compo-
nents of the extract, based on affinities for the medium
and solvent.
TPH E Gas chromatographic test for TPH extractable organic
compounds.
TPH V Gas chromatographic test for TPH volatile organic
compounds.
TPH-D(DRO) Gas chromatographic test for TPH diesel-range organics.
TPH-G(GRO) Gas chromatographic test for TPH gasoline-range organics.
Unresolved complex The thousands of compounds that a gas chromatograph
mixture (UCM) is unable to fully separate.
94

Volatile compounds “Volatile” is relative. It may mean (1) any compound
which will purge, (2) any compound which will elute
before the solvent peak (usually those < C
6
), or (3) any
compound which will not evaporate during a solvent
removal step.
Wax Waxes of petroleum origin consist primarily of normal
paraffins. Waxes of plant origin consist of esters of unsat-
urated fatty acids.
95

BIBLIOGRAPHY
American Petroleum Institute, Questions and Answers Publications.
American Public Health Association, Standard Methods for the Examination of Water
and Wastewater, 17th ed., Washington, DC, Publication Office, 1989.
ASTM Book of Standards. Section 5.02, American Society for Testing and Materials,
Philadelphia, PA, 1992.
ASTM Book of Standards. Section 5.01, American Society for Testing and Materials,
Philadelphia, PA, 1993.
ASTM Book of Standards. Section 5.03, American Society for Testing and Materials,
Philadelphia, PA, 1994.
Christensen, L.B., and Larsen, T.H., “Method for Determining the Age of Diesel
Oil Spills in the Soil,” Groundwater Monitoring Report, Fall 1993, pp. 142-149.
Coleman, W.E., et al., “The Identification and Measurement of Components in
Gasoline, Kerosene, and No. 2 Fuel Oil that Partition into the Aqueous Phase
After Mixing,” Arch. Environ. Contam. Toxicol. 13:171-174 (1984).
Corrosion Inhibitors, National Association of Corrosion Engineers, P.O. Box
218430, Houston, TX 77218 (1973).
Drews, A.W., Ed., ASTM Manual on Hydrocarbon Analysis, 5th ed., 1992.
Encyclopedia of Chemical Technology (New York: John Wiley & Sons, 1994).
EPA Draft Method 1664, n-Hexane Extractable Material and Silica Gel Treated n-
Hexane Extractable Material (SGT-HEM) by Extraction and Gravimetry (Oil
and Grease and Total Petroleum Hydrocarbons), 19 March 1994.
“EPA Method 418.1 Total Recoverable Petroleum Hydrocarbons by IR,”
Groundwater Analytical Technical Bulletin, Winter 1991/1992, Groundwater
Analytical Inc., Buzzards Bay, MA.
EPA Method 8240, Gas Chromatography/Mass Spectrometry for Volatile Organics,
Test Methods for Evaluating Solid Wastes, Volume 1B, 1986.
Gergel, W.C., “Lubricant Additive Chemistry,” The International Symposium of
Technical Additives and the Environment, Interlaken, Switzerland, May 22-25, 1984.
Gibbs, L.M., “Gasoline Additives - When and Why,” SAE Technical Paper Series,
International Fuels and Lubricants Meeting and Exposition, Tulsa, OK, October
22-26, 1990.
97

Hutcheson, M.S., D. Pedersen, N.D. Anastas, J. Fitzgerald and D. Silverman.
“Beyond TPH: Health-Based Evaluation of Petroleum Hydrocarbon Exposures,”
Regulatory Toxicology and Pharmacology, 24:85-101.
King, R.W., “Petroleum: Its Composition, Analysis and Processing,” Occupational
Medicine: State of the Art Reviews.
MA DEP, Office of Research and Standards (1994), “Interim Final Petroleum
Report: Development of Health-Based Alternative to the TPH Parameter,”
Prepared by ABB Environmental Services.
McGraw-Hill Encyclopedia of Science & Technology, Vols. 5,7,9,10,11 (New York:
McGraw-Hill Publishing Company, 1985).
McGraw-Hill Dictionary of Scientific and Technical Terms, 3rd ed. (New York: McGraw-
Hill Publishing Company, 1984).
McKetta, J.J., Industrial Products Handbook, Vols. 1 and 2 (New York: Marcel Dekker,
Inc., 1994).
Owen, K., and T. Coley, “Automotive Fuels Handbook,” Society of Automotive
Engineers, Inc., 1990.
Rhodes, I.A., et al., “Determination of Total Petroleum Hydrocarbons by Capillary
Gas Chromatography,” Shell Development Company, Environmental Analysis
Department, Westhollow Research Center, Houston, TX.
Rhodes, I.A., E.M. Hinojosa, D.A.Barker, and R.A. Poole, “Pitfalls Using
Conventional TPH Methods for Source Identification: A Case Study,” Proceedings
of the Seventeenth Annual Conference: EPA Analysis of Pollutants in the Environment,
Norfolk, VA, May 1994.
Schwerko, E.M., “TPH in Soil Primer,” BP America, Inc., 1993.
Senn, R.B., and M.S. Johnson, “Interpretation of Gas Chromatographic Data in
Subsurface Hydrocarbon Investigations,” Groundwater Monitoring Report, Winter
1987, pp. 58-63.
Speight, J.G., The Chemistry and Technology of Petroleum, 2nd ed. (New York: Marcel
Dekker, 1991).
Sullivan, B., and S. Johnson, “‘Oil’ You Need to Know About Crude — Implications
of TPH Data for Common Petroleum Products,” Soils, May 1993, pp. 8-13.
Swallow, K.C., N.S. Shrifin, and P.J. Doherty, “Hazardous Organic Compound
Analysis,” Environmental Science and Technology, 22(2):136-142 (1988).
Thomey, N., D. Bratberg, and C. Kalisz, “A Comparison of Methods for Measuring
Total Petroleum Hydrocarbons in Soil,” in Proceedings of the Petroleum Hydrocarbons
and Organic Chemicals in Groundwater: Prevention, Detection and Restoration,
National Water Well Association, November 15-17, 1989.
Wang, Z., M. Fingas, and G. Sergy, “Study of 22-Year-Old Arrow Oil Samples Using
Biomarker Compounds by GC/MS,” Environ. Sci. Technol., 28:1733-1746 (1994).
98
