
United States Government Accountability Office
Report to Congressional Committees
TECHNOLOGY ASSESSMENT
Regenerative Medicine
Therapeutic Applications, Challenges, and Policy
Options
July 2023
GAO-23-105430
The cover image displays a stylized representation of a human body, circled by icons representing key regenerative
medicine technologies.
Cover source: GAO; Anttoniart/derariad/greenvector/mariia/microone/texvector/stock.adobe.com (images). | GAO-
23-105430

United States Government Accountability Office
Highlights of GAO-23-105430, a report to
congressional committees
July 2023
TECHNOLOGY ASSESSMENT
Regenerative Medicine
Therapeutic Applications, Challenges, and
Policy Options
What GAO found
Regenerative medicine offers the hope of being able to restore or replace cell,
tissue, and organ functions affected by disease, injury, or aging. This may eventually
help manage or cure many conditions that are currently considered chronic,
untreatable, or terminal.
Examples of Diseases and Regenerative Medicine Therapies That Might Address Them
GAO identified many challenges that may affect the development and use of
regenerative medicine technologies and therapies including:
Challenges related to standardization. Standards are rules, conditions, guidelines,
or agreed-upon practices that are adopted within an industry to provide developers
with a common framework and promote consistency. Developing regenerative
medicine standards is challenging because these technologies and therapies are
complex and rapidly evolving. In addition, standards require consensus from
stakeholders, which may be difficult to obtain.
Challenges related to regulation. The Food and Drug Administration (FDA) ensures
the safety, efficacy, and security of human medical products in the U.S. through
regulation. Regenerative medicine faces challenges related to regulation, including
difficulty navigating a complex regulatory framework, uncertainty over which
regulatory pathway is most appropriate for certain emerging technologies and
therapies, and staffing shortages at FDA and collaborating agencies.
Challenges related to manufacturing. Manufacturing is the creation of products
from starting materials, in a way that is generally consistent and reproducible. It is a
key step for many emerging technologies and therapies, but the cells, tissues, and
organs used for regenerative medicine are complex and difficult to manufacture at
scale. Other challenges related to manufacturing include a lack of infrastructure and
difficulty ensuring quality and consistency.
View GAO-23-105430. For more information,
contact Karen L. Howard at (202) 512-6888
Why GAO did this study
Regenerative medicine represents a
paradigm shift in the medical field
because it aims to restore or
supplement function, rather than just
treating symptoms, and opens the
door for personalized therapies.
GAO conducted an assessment of
current and emerging regenerative
medicine technologies and
therapeutic applications. This report
examines (1) current and emerging
regenerative medicine technologies
and therapies and their potential
benefits, (2) challenges that hinder
their development and use, and (3)
policy options that could help
enhance benefits and mitigate
challenges associated with these
technologies and therapies.
GAO reviewed scientific and policy
literature and other key reports;
convened a 3-day expert meeting;
and interviewed subject matter
experts and stakeholder groups
including government agencies, such
as the Department of Health and
Human Services, non-government
organizations, industry, academia,
end user groups such as patient
groups. GAO is identifying policy
options in this report.

GAO developed 11 policy options that could help address the challenges or enhance the benefits of regenerative medicine.
These policy options are provided to inform policymakers of potential actions to address the policy challenges identified in this
technology assessment. They identify possible actions by policymakers, which include Congress, federal agencies, state and
local governments, academic and research institutions, and industry. Policymakers would need to consider the impacts these
new technologies will have on existing federal programs that are already strained. We suggested possible federal components
for the policy options. See tables 1-3 for a full list of the policy options, potential implementation approaches, and
opportunities and considerations.
Selected Policy Options to Mitigate Challenges Associated with Regenerative Medicine Technologies and Therapies
Selected policy option
Opportunities
Considerations
Invest in standards development.
(report p. 25)
This policy option could help address the
challenge that standards require consensus.
Could streamline standards development,
which may, in turn, accelerate innovation,
increase product safety and reliability,
accelerate regulatory review, and decrease
costs of regenerative medicine therapies.
Existing organizations may not include all
stakeholders, and stakeholders may
hesitate to accept standards created
without their input.
Industry stakeholders may hesitate to
adopt standards if they perceive it will
cost them a controlling position in the
market.
Standards should be appropriately
flexible to allow for innovation, while still
being detailed and specific enough to
support manufacturing of consistent,
quality products.
Provide opportunities for increased
interactions between regulatory
experts (at FDA or in industry) and
smaller companies, especially early in
the development process (report p. 31)
This policy option could help address the
lack of access to regulatory expertise.
May provide more timely advice and avoid
unnecessary delays or uncertainty by pursuing
the wrong regulatory pathways or submitting
data that do not meet regulatory
requirements.
May require additional resources to
bolster the workforce of regulatory
scientists at FDA or public-private
partnerships.
FDA may be limited in its ability to advise
companies early in the process so as not
to create a conflict of interest.
Consider whether changes to the
framework for evaluating combination
products and medical devices to
accommodate emerging technologies
and therapies may be necessary.
(report p. 32)
This policy option could help address
whether current regulatory pathways are
sufficient for emerging technologies and
therapies.
May encourage innovators, researchers, and
developers of new products to provide
valuable feedback to regulators.
Coordinating among stakeholders to
consider changes to regulatory pathways
may be time- and resource-intensive.
If such consideration leads to
recommended changes to the
framework, statutory and regulatory
changes may be necessary.
Provide more oversight and feedback
to suppliers to increase consistency in
starting materials (report p. 39)
This policy option could help address
inconsistency in starting materials for
manufacturing.
May accelerate manufacturing by reducing
variation in input materials.
May reduce the risk of failure during product
development.
Starting material suppliers may lack
incentives to follow standards if they
lead to higher costs.
Source: GAO. | GAO-23-105430
This is a work of the U.S. government and is not subject to copyright protection in the United States. The published
product may be reproduced and distributed in its entirety without further permission from GAO. However, because this
work may contain copyrighted images or other material, permission from the copyright holder may be necessary if you
wish to reproduce this material separately.
Regenerative Medicine GAO-23-105430 i
Table of Contents
Introduction ........................................................................................................................ 1
1 Background ...................................................................................................................... 3
1.1 Definition ........................................................................................................................ 3
1.2 How regenerative medicine works .................................................................................. 3
1.3 The development and licensure process for biologics ..................................................... 3
1.4 Advancements in regenerative medicine ........................................................................ 6
2 Current and Emerging Technologies in Regenerative Medicine ..................................... 7
2.1 Cell technologies ............................................................................................................. 8
2.2 Tissue technologies ....................................................................................................... 13
2.3 Organ technologies ....................................................................................................... 15
3 Challenges and Policy Options for Regenerative Medicine Technologies and
Therapies ........................................................................................................................... 21
3.1 Challenges related to standardization ........................................................................... 21
3.2 Challenges related to regulation ................................................................................... 26
3.3 Challenges related to manufacturing ............................................................................ 34
4 Agency and Expert Comments ....................................................................................... 41
Appendix I: Objectives, Scope, and Methodology ............................................................ 42
Objectives ........................................................................................................................... 42
Appendix II: Expert Participation ...................................................................................... 45
Appendix III: GAO Contact and Staff Acknowledgments .................................................. 46
Regenerative Medicine GAO-23-105430 ii
Tables
Table 1: Policy options for regenerative medicine standardization ................................. 25
Table 2: Policy options for regenerative medicine regulation ......................................... 31
Table 3: Policy options for regenerative medicine manufacturing .................................. 39
Figures
Figure 1: Conventional development and licensure process for regenerative medicine
products .............................................................................................................................. 5
Figure 2: Levels of complexity in cells, tissues, and organs ................................................ 8
Figure 3: Scaffold de- and recellularization of a liver ....................................................... 15
Figure 4: An engineered bladder using a patient’s cells and a biodegradable scaffold ... 20
Figure 5: Comparison of potential centralized and distributed manufacturing models .. 36
Regenerative Medicine GAO-23-105430 iii
Abbreviations
AMD
age-related macular degeneration
CAR T Cell
chimeric antigen receptor T cell
CQA
critical quality attribute
CBER
Center for Biologics Evaluation and Research
CDER
Center for Drug Evaluation and Research
CDRH
Center for Devices and Radiological Health
FDA
Food and Drug Administration
NIST
National Institute of Standards and Technology
RMAT
regenerative medicine advanced therapy
SCB
Standards Coordinating Body for Gene, Cell, and Regenerative Medicines
and Cell-Based Drug Discovery

Regenerative Medicine GAO-23-105430 1
441 G St. N.W.
Washington, DC 20548
Introduction
July 13, 2023
The Honorable Bernard Sanders
Chair
The Honorable Bill Cassidy, M.D.
Ranking Member
Committee on Health, Education, Labor, and Pensions
United States Senate
The Honorable Frank D. Lucas
Chair
The Honorable Zoe Lofgren
Ranking Member
Committee on Science, Space, and Technology
House of Representatives
Regenerative medicine technologies offer the hope of creating therapeutic products that restore
cell, tissue, and organ functions affected by disease, injury, or aging. These technologies
represent a paradigm shift in the medical field, away from developing therapies that treat
symptoms and toward creating products that cure the underlying disease or restore function.
They also open the door to personalized therapies that use an individual’s own genes or cells,
sometimes engineered to replace or augment their functions. Currently, these technologies are
being used to create life-saving therapies for broad categories of diseases, which may help
Americans with diabetes (accounting for one-quarter of all U.S. health care costs), cancer (about
1.7 million new cases annually), non-fatal fall injuries (about 8 million cases in 2018), or age-
related macular degeneration (AMD) (about 20 million cases overall as of 2019).
1
In addition,
regenerative medicine may one day offer relief to the approximately 104,000 individuals in need
of an organ transplant who are on a waiting list that far exceeds availability.
2
1
Centers for Disease Control and Prevention, Cancer Data and Statistics, https://www.cdc.gov/cancer/dcpc/data/, accessed Mar. 17,
2023. Briana Moreland et al. Trends in Nonfatal Falls and Fall-Related Injuries Among Adults Aged ≥65 Years — United States, 2012–
2018. MMWR Morb Mortal Wkly Rep 2020;69:875–881. http://doi.org/10.15585/mmwr.mm6927a5. Centers for Disease Control
and Prevention, Prevalence of Age-Related Macular Degeneration (AMD), https://www.cdc.gov/visionhealth/vehss/estimates/amd-
prevalence.html, accessed Mar. 17, 2023.
2
The Health Resources and Services Administration website shows 104,200 individuals were on the organ transplant waiting list as of
March 2023. https://www.organdonor.gov/learn/organ-donation-statistics, accessed Mar. 28, 2023.

Regenerative Medicine GAO-23-105430 2
GAO has done prior work on funding streams, workforce, and education for regenerative
medicine and the known problems within the organ transplant system.
3
We prepared this report
under the authority of the Comptroller General in light of congressional interest in the potential
of this field. This report examines:
(1) current and emerging regenerative medicine technologies and therapies and their
potential benefits,
(2) challenges that hinder the development and use of regenerative medicine
technologies and therapies, and
(3) policy options that could help enhance benefits and mitigate challenges associated
with these technologies and therapies.
To address these objectives, we conducted a literature search; interviewed officials and
representatives from government, industry, academia, and end user groups such as patient
groups; and convened a 3-day expert meeting. See appendix I for the full objectives, scope,
and methodology used in this report and appendix II for the list of participants in our
expert meeting.
We conducted our work from September 2021 through July 2023 in accordance with all sections
of GAO’s Quality Assurance Framework that are relevant to technology assessments. The
framework requires that we plan and perform the engagement to obtain sufficient and
appropriate evidence to meet our stated objectives and to discuss any limitations to our work.
We believe that the information and data obtained, and the analysis conducted, provide a
reasonable basis for the findings and conclusions in this product.
3
GAO, Regenerative Medicine and Advanced Therapies: Information on Workforce and Education, GAO-23-106030 (Washington,
D.C.: Mar. 23, 2023); Organ Transplants: Changes in Allocation Policies for Donated Livers and Lungs, GAO-21-70 (Washington, D.C.:
Oct. 16, 2020); Regenerative Medicine: Federal Investment, Information Sharing, and Challenges in an Evolving Field, GAO-15-553
(Washington, D.C.: June 23, 2015).

Regenerative Medicine GAO-23-105430 3
1 Background
1.1 Definition
Regenerative medicine refers to a general
approach to restore, replace, or recreate
cells, tissues, or organs to treat or mitigate
disease.
4
Under the Federal Food, Drug, and
Cosmetic Act, the Food and Drug
Administration (FDA) regulates regenerative
medicine products, which include cell
therapies, therapeutic tissue engineering
products, combination products using such
therapies or products, some gene therapy
products, and certain human cell and tissue
products.
5
1.2 How regenerative medicine
works
Regenerative medicine aims to develop new
therapies that offer benefits beyond those
offered by existing medical treatments.
These therapies can be highly personalized
and may eventually help manage or cure
many conditions that are currently
considered chronic, untreatable, or
terminal. These include heart disease,
diabetes, cancer, and sickle cell disease, as
well as severe burns and certain types of
bone fractures.
Regenerative medicine works by harnessing
the body’s own healing ability to restore
lost function, to establish normal function
4
See 21 U.S.C. § 356(g)(8).
5
Certain regenerative medicine products may be eligible for
regenerative medicine advanced therapy (RMAT)
that was absent at birth, or to augment
natural function to fight a disease. There is
a wide range of technologies available in
the field. For example, some researchers
are using gene editing technology to correct
genetic defects or introduce new healing
capabilities for diseases such as sickle cell
disease. Another tool is the use of
implanted materials that, unlike existing
medical implants, interact with the body to
encourage healing. Yet another is tissue
engineering, the practice of combining
materials, cells, and biologically active
molecules into functional tissues. These
tools can often be used on or in
combination with patients’ own cells, which
could bring additional benefits. For
example, the use of a patient’s own cells to
create a personalized organ could
transform organ transplantation by
alleviating donor organ shortages and
eliminating organ rejection—a reaction to
foreign biological material that requires
transplant patients to take
immunosuppressive drugs for the rest of
their lives.
1.3 The development and licensure
process for biologics
Biologics, a category that includes
regenerative medicine products, are a
diverse group of products regulated by
designation, which provides drug sponsors with certain
benefits, such as expedited review. See 21 U.S.C. § 356(g).

Regenerative Medicine GAO-23-105430 4
FDA.
6
FDA is responsible for the safety,
efficacy, and security of human medical
products marketed in the U.S., which for
biologics, includes premarket review and
approval of a biologics license application.
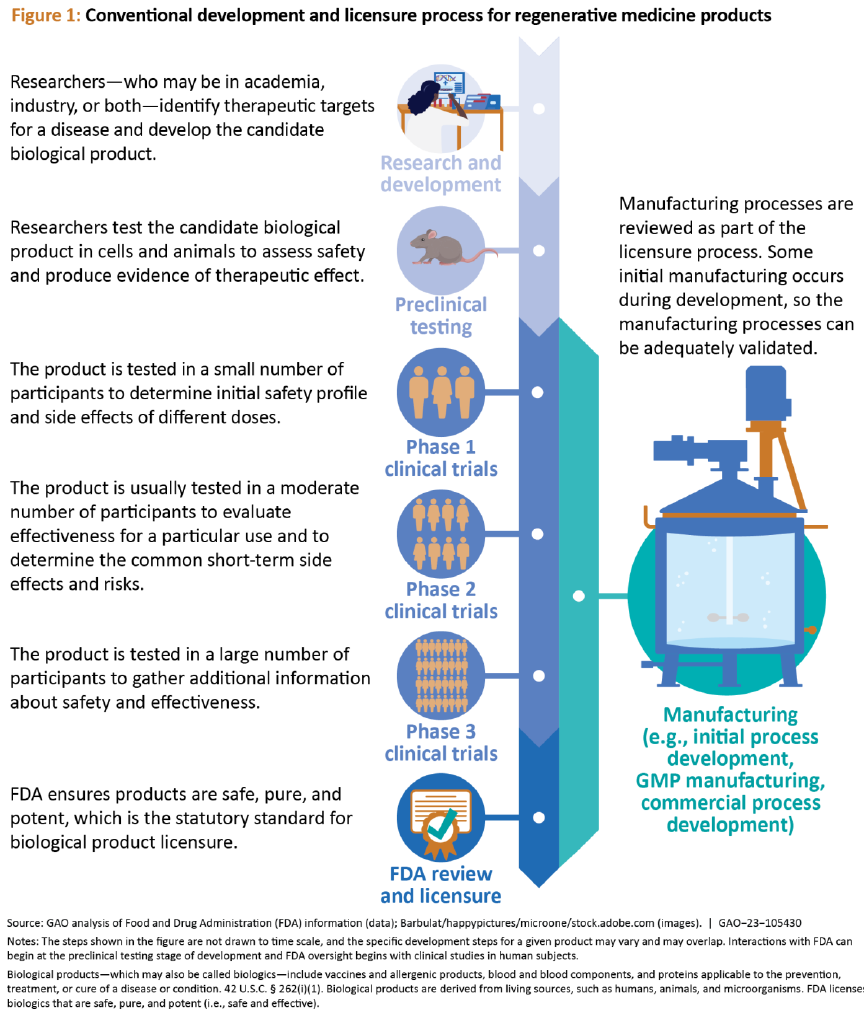
Figure 1 shows the conventional process for
developing and licensing regenerative
medicine products.
Depending on the medical product type,
different FDA centers may handle the
review process: the Center for Drug
Evaluation and Research (CDER) (which
regulates drugs and certain biologics), the
Center for Biologics Evaluation and
Research (CBER) (which regulates most
biologics), and the Center for Devices and
Radiological Health (CDRH) (which regulates
devices). Agency officials told us that
regenerative medicine products are
generally under the purview of CBER. For
combination products—such as those that
combine two or more regulated products
(e.g., a biologic and a device)—the center
with primary jurisdiction over the product’s
primary mode of action will review and
regulate the product.
7
6
Biological products—which may also be called biologics—
include vaccines and allergenic products, blood and blood
components, and proteins applicable to the prevention,
treatment, or cure of a disease or condition. 42 U.S.C. §
262(i)(1). Biologics are derived from living sources, such as
humans, animals, and microorganisms. FDA licenses
biologics that are safe, pure, and potent (i.e., safe and
effective).
7
The primary mode of action is the single mode of action of
a combination product that provides the most important
therapeutic action of the combination product. The most
important therapeutic action is the mode of action expected
to make the greatest contribution to the overall intended
therapeutic effect of the combination product. 21 C.F.R. §
3.2(m) (2022). The Office of Combination Products assigns
combination products to FDA’s medical product centers for
review, and coordinates reviews involving more than one
FDA center.
Regenerative Medicine GAO-23-105430 5

Regenerative Medicine GAO-23-105430 6
1.4 Advancements in regenerative
medicine
FDA first licensed a tissue-engineered
product in 1998—a skin graft for the
treatment of a form of skin ulcers.
8
Since
then, technological advances have
increased steadily, and the number of
investigational new drug applications for
regenerative medicine products, as well as
the number of products in clinical trials
continues to grow.
9
These applications
include cell therapy to cure blood cancers
and gene therapy to cure sickle cell disease.
Further, researchers have successfully
grown whole organs such as livers
and bladders.
Despite these advances, the number of
regenerative medicine products licensed for
use in humans remains small. Many
regenerative medicine products are
considered more complex than certain
other biologics, such as monoclonal
antibodies. Unlike drugs, cells and tissues
are living, constantly changing, and variable
from person to person. This fact underpins
many of the challenges in the field, which
we describe in chapter 3.
Recent laws may help accelerate medical
product development, bringing new
innovations and advances to patients more
quickly and efficiently. For example, the
21st Century Cures Act created an
expedited process for FDA evaluation of
certain regenerative medicine therapies,
known as the regenerative medicine
advanced therapeutic (RMAT)
designation.
10
Chapter 2 of this report discusses the
current and emerging technologies in
regenerative medicine, including cell, tissue,
and organ technologies that may be used to
develop therapeutic products. Chapter 3
discusses the challenges that researchers
and developers face in developing and
bringing regenerative medicine products to
market. In chapter 3, we also present policy
options that may help address
these challenges.
8
FDA CDRH, Summary of Safety and Effectiveness Data,
https://www.accessdata.fda.gov/cdrh_docs/pdf/P950032S0
16b.pdf, 7, accessed Mar. 27, 2023.
9
A drug sponsor may not conduct human clinical trials until
it has submitted an investigational new drug application to
FDA. Once submitted, the sponsor may begin clinical trials
after 30 days unless FDA issues a clinical hold. See 21 C.F.R. §
312.40 (2022).
10
Pub. L. No. 114-255, § 3036, 130 Stat. 1033, 1104 (2016)
(codified at 21 U.S.C. § 356(g)). FDA is required to designate
a drug as a regenerative medicine advanced therapy if (1)
the drug is a cell therapy, therapeutic tissue engineering
product, human cell or tissue product, or combination
product (with certain exceptions); (2) the drug is intended to
treat, modify, reverse, or cure a serious or life-threatening
disease or condition; and (3) preliminary clinical evidence
indicates that the drug has the potential to address unmet
medical needs for such disease or condition. Once an RMAT
designation has been made, FDA is required to facilitate an
efficient development program for and expedite review of
the drug. RMAT designation includes the benefits of certain
other expedited programs, and early interactions with FDA
may be used to discuss potential surrogate or intermediate
endpoints to support accelerated approval. See 21 U.S.C. §
356(g).

Regenerative Medicine GAO-23-105430 7
2 Current and Emerging Technologies in Regenerative Medicine
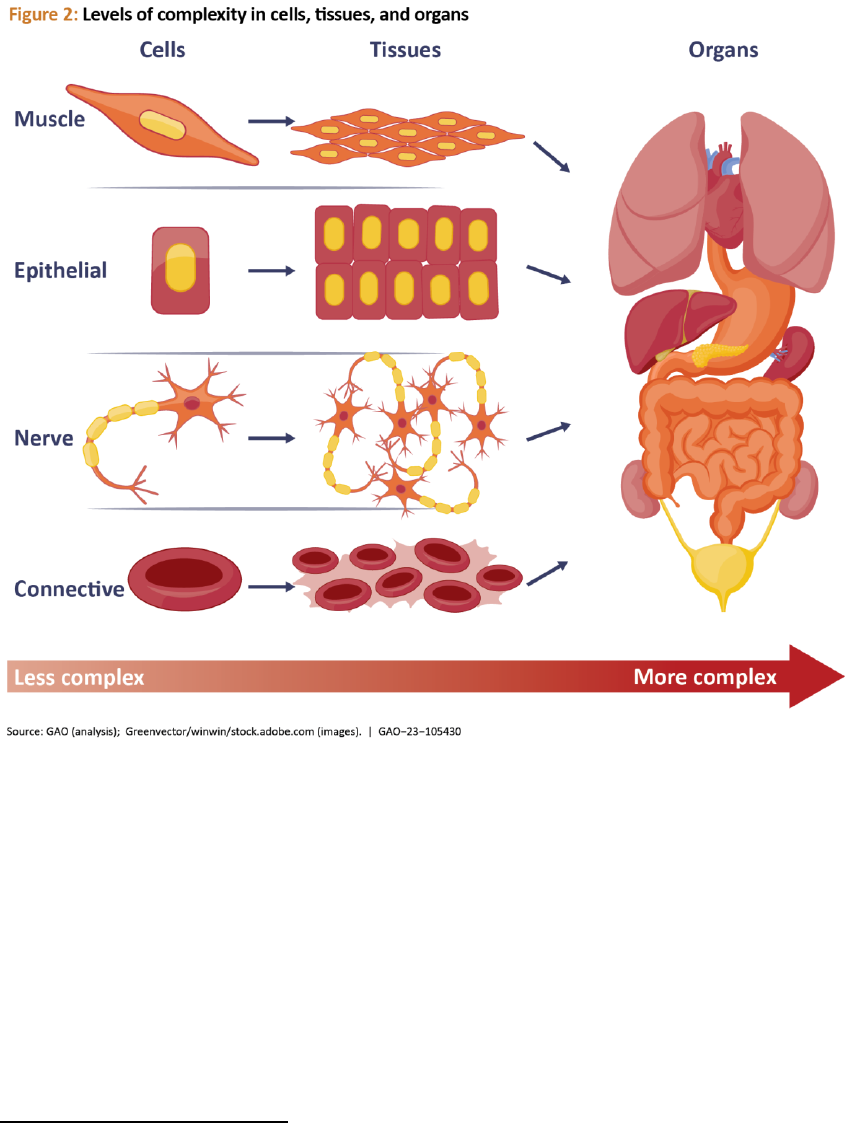
Regenerative medicine technologies can be
grouped in various ways including broad
categories such as cells, tissues, and organs,
which can be used to develop therapeutic
products. These vary in complexity
according to their level of structural
organization.
11
A cell is a self-sustainable
unit that can replicate itself and carry on all
the metabolic processes essential for life.
Tissues are groups of cells that function
together as a unit. For example, epithelial
tissue lines the various passages inside the
body such as the intestinal lining, and also
11
For the purposes of this report, technologies are grouped
into broad categories that aim to regenerate or restore cells,
tissues, and organs. FDA uses the term “cell and gene
therapy products” to describe a wide range of products.
makes up the skin. Organs are collections of
several different tissues arranged to
perform a special function in the body. The
human heart, for example, contains cardiac
muscle tissue, connective tissue (which
holds the muscle tissue together), epithelial
tissue (which creates the lining of the
heart), nerve tissues, and specialized
pacemaker cells, which coordinate the
heartbeat. The level of structural
organization increases moving from cells to
tissues to organs, leading to technologies
with increasing engineering complexity (see
fig. 2).
Gene therapy products are biologics, as the term is defined
under 42 U.S.C. § 262(i)(1). While human gene therapy
products may include ex vivo modified cells, FDA
distinguishes between cellular and gene therapies.
Regenerative Medicine GAO-23-105430 8
2.1 Cell technologies
Cells are the smallest units of life and make
up all living organisms. Each cell has a full
set of genetic material (i.e., a genome) that
provides the instructions needed to
perform essential processes and reproduce.
Cell-based regenerative medicine
technologies may be used to develop cures
for a variety of diseases and can use either
12
The process by which a cell becomes specialized in order
to perform a specific function is called ‘differentiation.’
When cells differentiate, certain genes are turned on or off
and this determines what type of cell will result.
cells from a patient’s own body or cells
from a donor as the starting material for
therapy. Regenerative medicine
technologies may use specialized or
unspecialized cells. Specialized cells are
those that have undergone genetic changes
to become a specific type of cell, such as a
red or white blood cell.
12
Unspecialized
cells, which are known as stem cells and
found in both embryos and adults, have not
yet undergone these changes and have the

Regenerative Medicine GAO-23-105430 9
ability to become different types of cells.
Finally, regenerative medicine technologies
may incorporate gene-editing techniques to
produce gene-edited cells.
The following describes current and
potential cell-based therapies. We group
them into therapies based on stem cells and
those based on gene-edited cells, although
some therapies use stem cells that have
also been gene-edited.
Stem cell therapies. Stem cells have been
used to replace damaged cells and restore
or improve bodily functions since the first
bone marrow transplant more than 60
years ago (bone marrow makes stem cells).
Today, there are several types of stem cell
transplants. For example, hematopoietic
stem cell transplants provide a person with
a blood disorder, such as anemia or cancer,
with an infusion of stem cells that restores
their ability to produce blood cells.
13
Depending on the circumstances, the stem
cells may be obtained from the patient or a
donor and may be derived from bone
marrow, peripheral blood, umbilical cord
blood, or other sources. Stem cells have
also been used in certain types of tissue
grafts for patients with corneal eye diseases
and skin grafts for burn victims.
14
Stem cell therapies have the potential to
cure numerous diseases and injuries. Initial
13
Hematopoiesis is the term for blood cell production. The
body continually makes new blood cells to replace old ones
to supply oxygen to the tissues (red blood cells), fight
infection (white blood cells), and clot the blood after injury
(platelets). Stem cell transplant for cancer may help to
restore normal stem cells after chemotherapy or radiation,
or it may act against cancers like leukemia or myeloma.
14
For a more detailed explanation of how stem cells are
used in tissue grafts for eye and skin regeneration, see de
research in the 1950s and 1960s used
embryonic stem cells from mice, as they are
more flexible and have the natural ability to
turn into any type of cell. However,
controversies around the use of human
embryonic stem cells turned researchers’
focus toward applying gene-editing
techniques to specialized cells and adult
stem cells (see text box).
Gene-edited cell therapies. Gene-edited
cells have been manipulated using a gene
Araujo, Aline Lütz, and José Álvaro Pereira Gomes. “Corneal
stem cells and tissue engineering: Current advances and
future perspectives.” World journal of stem cells, vol. 7, 5
(2015): 806-14. https://doi.org/10.4252/wjsc.v7.i5.806. And
Chen, Ming et al. “Stem cells for skin tissue engineering and
wound healing.” Critical reviews in biomedical engineering,
vol. 37, 4-5 (2009): 399-421.
https://doi.org/10.1615/critrevbiomedeng.v37.i4-5.50.
Embryonic stem cell concerns and the discovery of
induced pluripotent stem cells
Embryonic stem cells come from a human embryo and
their use has raised ethical concerns. In January 1996,
federal law prohibited the use of federal funds on
research that created or destroyed human embryos.
a
This
policy limited some research on embryonic stem cells, and
led scientists to search for alternative stem cell sources.
In 2006, researchers identified conditions that allowed
adult human cells to revert to a state similar to an
embryonic stem cell. In 2007, researchers developed the
first human cells of this kind, known as induced
pluripotent stem cells.
b
Similar to embryonic stem cells,
induced pluripotent stem cells can change into all types of
cells in the body. These cells can provide a replacement
for embryonic stem cells. They may be derived from a
patient’s own cells, offering the benefit of avoiding
rejection by the host immune system.
Source: GAO. | GAO-23-105430
a
Balanced Budget Downpayment Act, I, Pub. L. No. 104-99, § 128, 110 Stat.
26, 34 (1996). The parameters on fetal research, transplantation of fetal
tissue, and prohibitions regarding fetal tissue are governed by the provisions
of 42 U.S.C. §§ 289g-289g-2.
b
See Kazutoshi Takahashi et al. “Induction of pluripotent stem cells from adult
human fibroblasts by defined factors.” Cell, vol. 131, 5 (2007): 861-72.
https://doi.org/10.1016/j.cell.2007.11.019.

Regenerative Medicine GAO-23-105430 10
editing technology, such as CRISPR, to alter
a gene that codes for a particular protein.
15
These changes can restore cellular functions
or give cells new functions, such as the
potential to fight disease. Gene editing can
be used on specialized cells or stem cells.
For example, chimeric antigen receptor
(CAR) T cells are gene-edited versions of a
patient’s own immune cells that target and
kill certain types of cancer cells in their
body (see vignette 1).
16
Similarly, gene-edited stem cell therapies
are being used to treat sickle cell disease,
an inherited blood disorder that causes
sickle-shaped red blood cells (see vignette
15
CRISPR and other gene editing technologies can delete,
insert, replace, or modify parts of a cell’s DNA. DNA is a
molecule that stores hereditary information in humans and
other organisms. For more information on gene editing
technologies and CRISPR, see GAO, Science & Tech Spotlight:
CRISPR Gene Editing. GAO-20-478SP (Washington, D.C.: Apr.
7, 2020).
2). The combination of gene editing and
stem cells could help researchers achieve
therapy breakthroughs for a variety of
diseases. This includes severe combined
immunodeficiency, a group of hereditary
diseases that severely compromises or
destroys the immune system;
leukodystrophies, which are rare,
degenerative diseases of the nervous
system; and junctional epidermolysis
bullosa, a group of genetic conditions that
cause the skin to be very fragile and to
blister easily.
16
T cells, also known as T lymphocytes or thymocytes, are
part of the immune system and develop from stem cells in
the bone marrow. They help protect the body from infection
and may help fight cancer. CAR T cells are modified versions
of T cells.
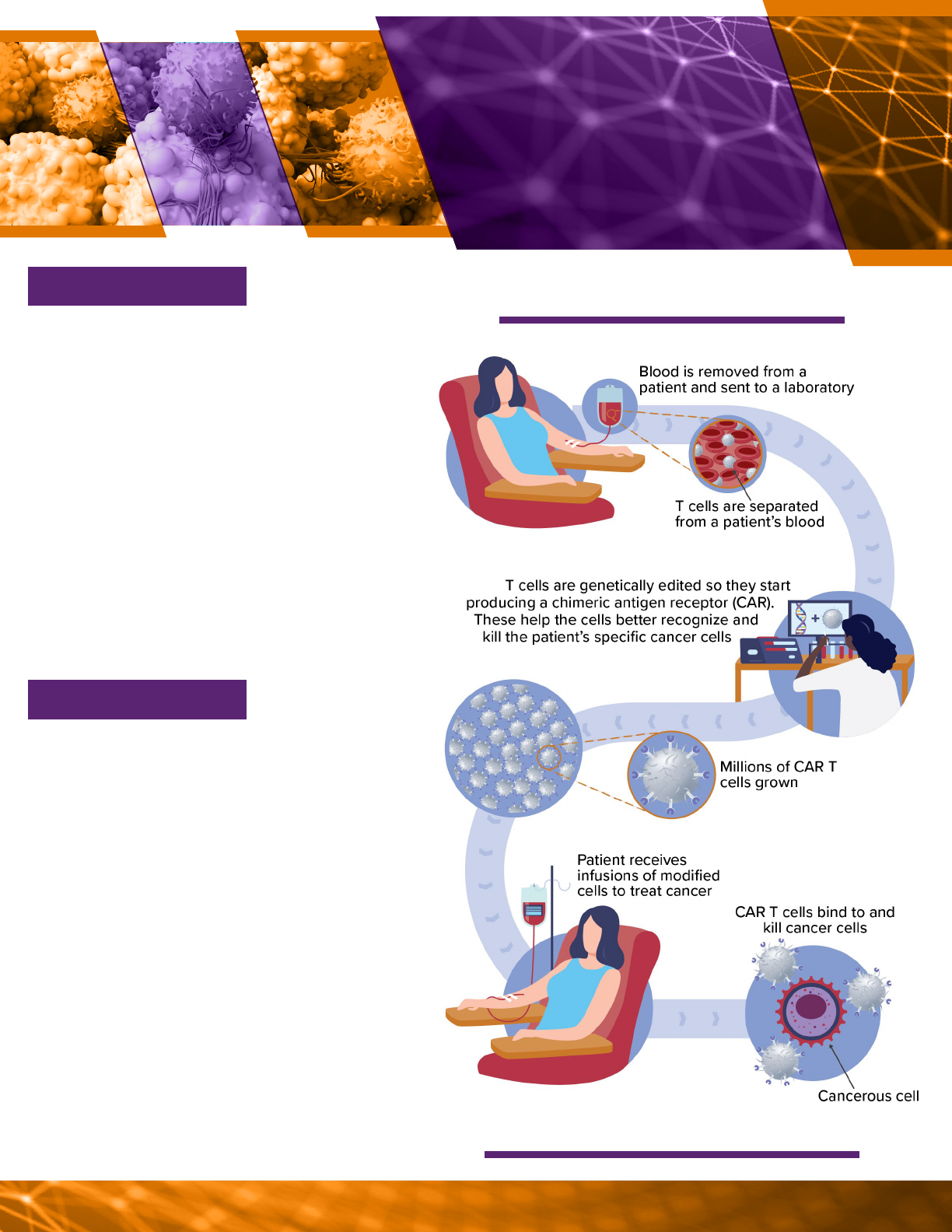
Source: GAO (analysis); Designua/greenvector/microone/topvectors/
stock.adobe.com (images). | GAO-23-105430
A possible process for making CAR T cells.
Cancer occurs when cells grow uncontrolla-
bly. It is among the leading causes of death
worldwide, and an estimated 1.7 million
Americans are diagnosed with cancer every
year—about 186,000 of them with leukemia,
lymphoma, or myeloma. These blood cancers
are caused by excessive production of white
blood cells in the bone marrow. Patients un-
dergoing treatment for cancer often receive
chemotherapy or radiation, but recurrence is
common. Chimeric antigen receptor (CAR) T
cells are a therapy alternative for patients for
whom standard treatment is not eective, or
whose cancer returns after initial treatment.
CAR T cell therapies have emerged as one of
the major breakthroughs in cancer therapies
over the last decade. The first CAR T cell
therapy received FDA licensure in 2017. As
of March 2022, there are at least six licensed
therapies for various types of blood cancers.
Researchers are developing new CAR T
therapies for other types of cancers. For
example, some early studies have shown that
CAR T cells may be able to treat solid tumors,
such as glioblastoma, which is an aggressive
type of cancer that can occur in the brain or
spinal cord. Researchers are also exploring
the use of donor cells for CAR T therapies,
which may enable larger-scale manufacturing.
WHAT’S NEXT?
WHAT IS IT?
CHIMERIC ANTIGEN
RECEPTOR T CELLS
AS A THERAPY FOR
CANCER
VIGNET TE 1
Source: Design cells/solvod/stock.adobe.com (images). | GAO-23-105430
Regenerative Medicine GAO-23-105430 11
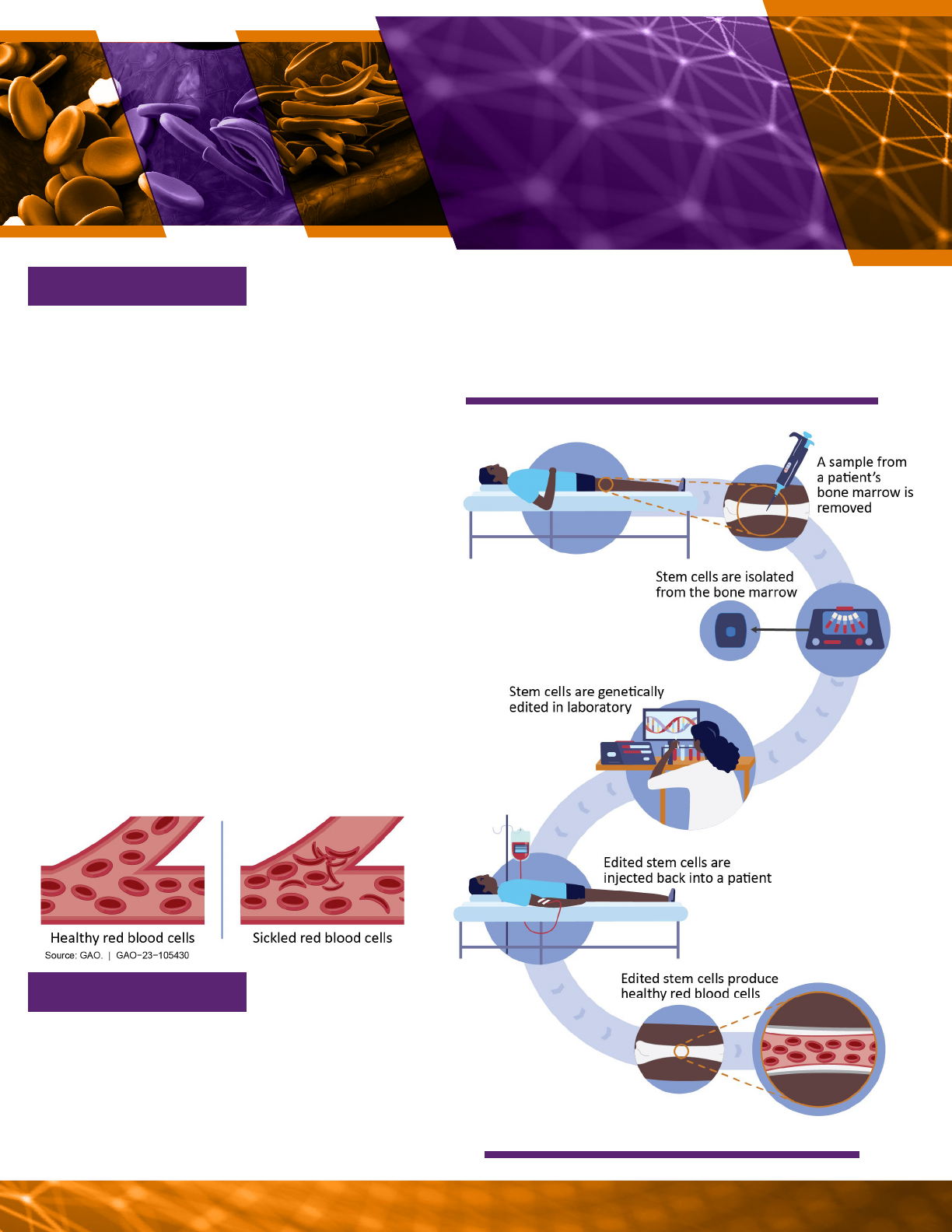
Regenerative Medicine GAO-23-105430 12
Source: GAO (analysis); Mariia/microone/stock.adobe.com (images). |
GAO-23-105430
A possible process for genetically editing stem cells.
disorder called beta-thalassemia. Other applications of this
technology—for sickle cell disease and other diseases, such
as diabetes—are being studied in phase 1 and 2 clinical trials.
GENE
-
EDITED STEM
CELLS AS A THERAPY
FOR SICKLE CELL
DISEASE
VIGNETTE 2
Source: tussika/solvod/stock.adobe.com (images). | GAO-23-105430
WHAT IS IT?
Sickle cell disease is a group of inherited
genetic disorders caused by an abnormal he-
moglobin gene. This gene causes red blood
cells to stick together and take on a rigid sickle
shape rather than the flexible round shape
found in healthy cells. Approximately 100,000
Americans are aected by sickle cell disease,
including approximately one in 365 African
Americans. Sickled cells can cause a broad
range of symptoms, including pain, stroke,
and organ damage. Current patient care is
primarily limited to relieving symptoms rather
than treating the disease. Some patients with
sickle cell disease may receive blood transfu-
sions or bone marrow transplants, but these
therapies have risks. Red blood cells come
from bone marrow stem cells, so genetically
editing stem cells can correct a patient’s he-
moglobin gene and lead to the production of
healthy red blood cells.
Genetically edited stem cells have significant
potential for treating hereditary and rare
diseases, according to experts. In August
2022, FDA licensed the first gene-edit-
ed stem cell treatment for a related blood
WHAT’S NEXT?
Regenerative Medicine GAO-23-105430 13
2.2 Tissue technologies
Tissue technologies for regenerative medicine
combine cells and biocompatible materials
into a single product. By combining these
materials with cells, tissue technologies help
cells stay at a specific location in the body,
provide structural support, and enable more
targeted therapeutic approaches.
The following describes two categories of
tissue technologies that may have therapeutic
applications:
Biocompatible materials. Biocompatible
materials come from natural or artificial
sources and serve as structural scaffolds.
When implanted into a patient, they can be
used to support or replace damaged tissues.
Certain materials, such as metals, ceramics,
plastic, or glass, have been used extensively
as surgical implants and scaffolds because
they replace the function of tissue and are
not biologically active—meaning they
typically do not actively interact with a
patient’s body. Biomaterials under
development for regenerative medicine
technologies—such as hydrogels—differ from
those currently used in surgical implants
because they are not inert and are designed
for cells to attach or interact with them to
actively facilitate healing responses. While
these materials have the potential to
significantly advance regenerative medicine,
there are limitations. For example, new
applications of biologically active or
regenerative materials will require much
closer monitoring and testing to ensure
patient safety because they do not have the
well-established performance records of inert
materials.
Combination products. Combination products
are products made up of two or more
components regulated by FDA. For example, a
tissue-engineered product containing both
living cells and biocompatible materials is
classified as a combination product because it
has elements of both a biologic and device.
Combination products may address certain
age-related conditions that can cause
structural and functional changes in the cells
and tissues. For example, a retinal implant
that combines a patient’s cells with a
biodegradable scaffold to create a
combination product may cure advanced dry
age-related macular degeneration (AMD), an
eye disease that can blur the central part of a
person’s vision (see vignette 3).
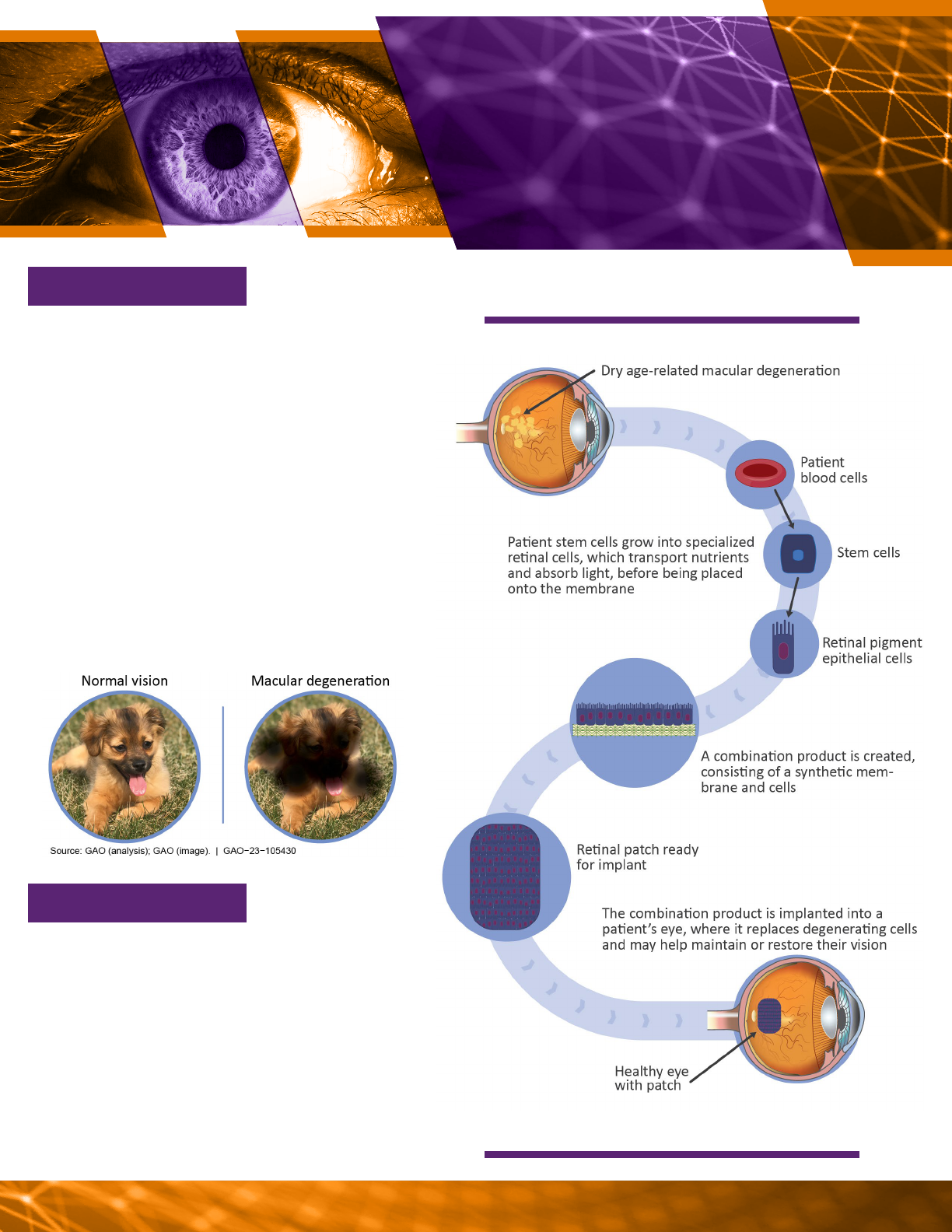
Regenerative Medicine GAO-23-105430 14
Source: GAO (analysis); Greenvector/pattarawit/stock.adobe.com (images). |
GAO-23-105430
A possible process for making retinal implants.
RETINAL IMPLANTS AS A
THERAPY FOR DRY AGE-
RELATED MACULAR
DEGENERATION
VIGNETTE 3
Source: Firefighter Montreal/solvod/stock.adobe.com (images). | GAO-23-105430
WHAT IS IT?
Dry age-related macular degeneration (AMD)
is an eye disease caused by damage to a
person’s retina as they age. Approximately
20 million Americans have AMD, more than
1.7 million of whom have an advanced form
of the disease that results in vision loss. Such
vision loss makes it hard to do everyday tasks,
including seeing faces, reading, driving, or
working around the house. There are currently
no eective therapies. Retinal implants—a
patch made from a patient’s cells and a
synthetic scaold—are being developed with
the hope of providing the first therapy for this
type of vision loss.
At least three dierent stem-cell-based
therapies for AMD are in phase 1 and 2 clinical
trials. Further developments in tissue engi-
neering may pave the way for other combina-
tion products made from a patient’s own cells.
Researchers are exploring tissue engineering
for other conditions, but it is dicult to predict
the future direction of this technology given its
early development stage.
WHAT’S NEXT?
Regenerative Medicine GAO-23-105430 15
2.3 Organ technologies
Organ technologies, such as artificial hearts
and kidneys, can have more complex
structures and functions than cell or tissue
technologies. They combine multiple cell and
tissue types to create complex 3D structures.
New strategies will be required to support
these technologies.
Some technologies under development for
potential therapeutic application include the
following:
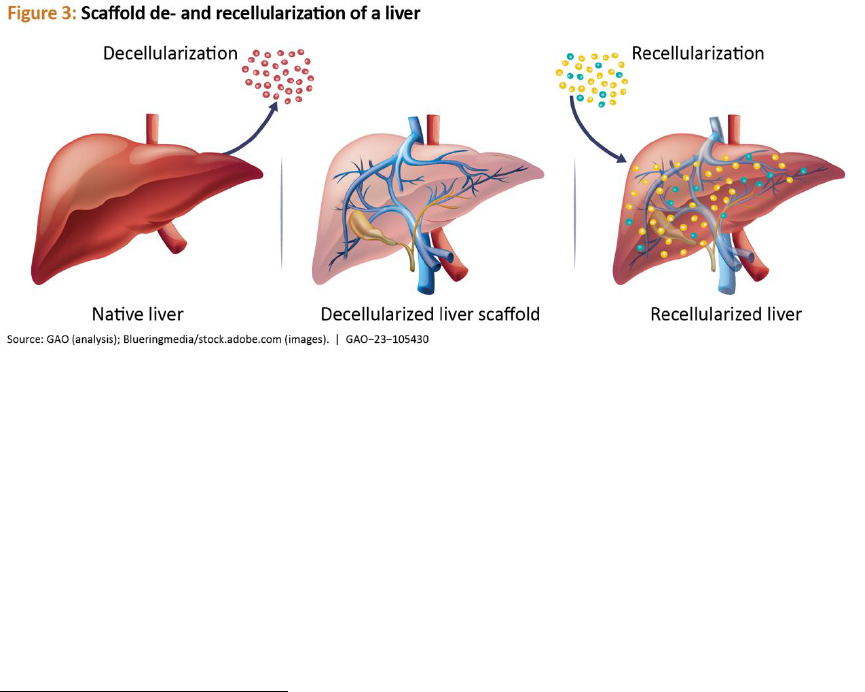
Scaffold de- and recellularization. Scaffold
decellularization removes cells from tissues or
organs and leaves behind the non-cellular
portion of a tissue (i.e., scaffold) which mainly
provides physical support. Recellularization
adds new cells from a patient or other
external source to the scaffold, where those
cells will attach and grow. Patients needing
organ transplants may benefit from the use of
this technology once it is more developed. For
example, a pig liver can be decellularized and
the resulting scaffold may be repopulated
with patient-derived cells, which makes it less
likely that the new liver would be rejected
(see fig. 3).
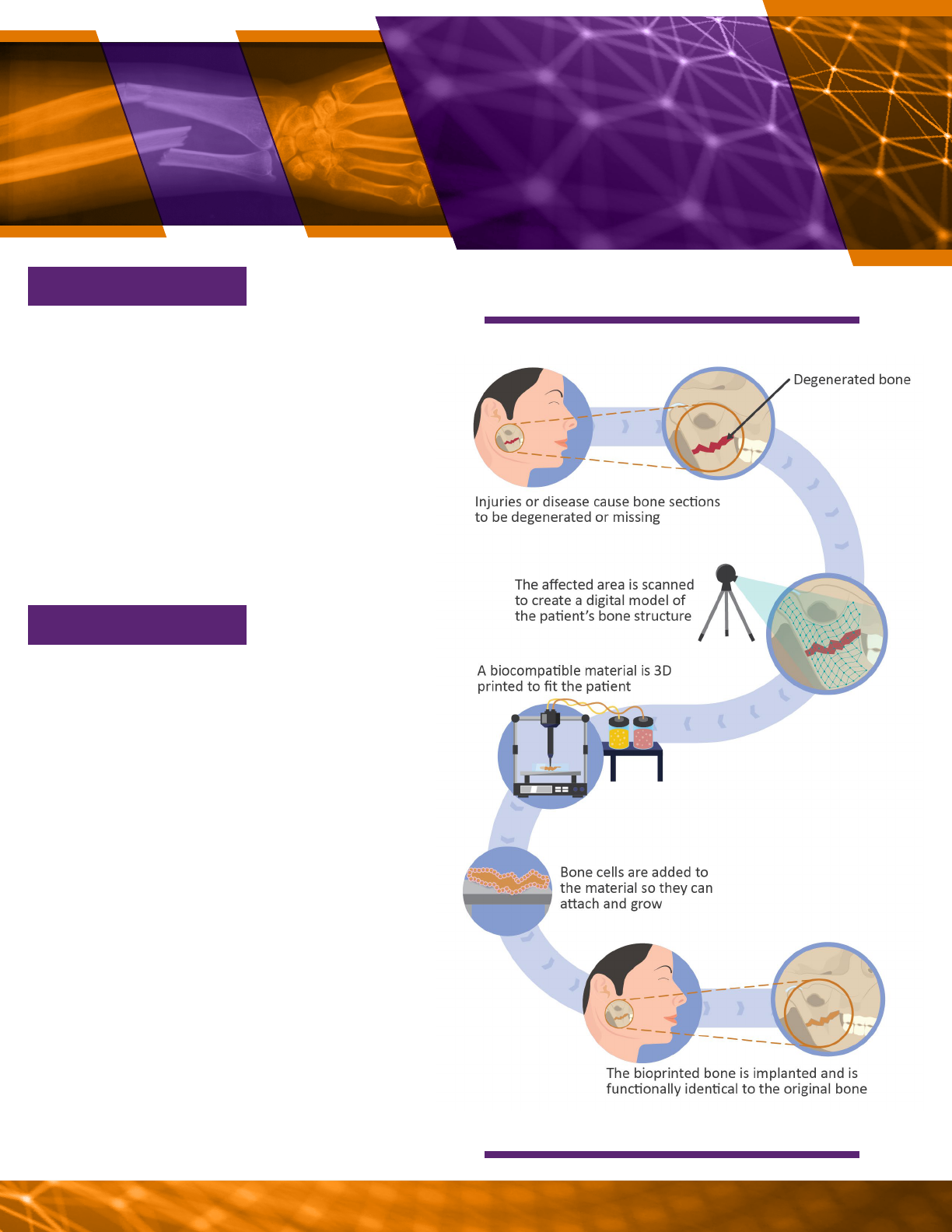
3D bioprinting. 3D bioprinting uses 3D
printing techniques to create implantable
structures. The material used as ink for the 3D
printer can contain cells, or cells can be added
after printing is complete. Researchers have
successfully implanted 3D printed bone and
muscle structures into animals. Additionally,
in June 2022, a human patient received a 3D
printed ear implant as part of a clinical trial.
17
17
See ClinicalTrials.gov, AuriNovo for Auricular Reconstruction,
https://clinicaltrials.gov/ct2/show/NCT04399239, accessed
Mar. 28, 2023.
These advances highlight the potential
application of 3D bioprinted technologies, but
applications that allow for the treatment of
human disease are still under development.
For example, researchers are pursuing 3D
printed tissues to cure bone defects or
injuries (see vignette 4).
Regenerative Medicine GAO-23-105430 16
Source: GAO (Analysis). Derariad /rumruay/ stock.adobe.com (images). |
GAO-23-105430
A possible process for bioprinting bone material.
Injuries and accidents can cause bone
fractures. Between 11 million and 15 million
bone fractures occur in the U.S. every year, of
which more than 1 million fail to heal properly.
Current therapies may use transplanted
tissues or inorganic materials, but neither of
these fully restores functionality. Bioprinted
bones could combine a 3D printed biocom-
patible material with a patient’s own bone
cells to create customized replacements for
damaged bone.
3D bioprinted bone replacements are still
in research and development. No bone
construct has been made by combining
tissue engineering and 3D bioprinting, but
studies have been done in animals. Further
progress requires research into creating
blood vessels in implanted materials and
developing stronger, more flexible materials,
among other areas. Additionally, a report from
the Pew Charitable Trusts published in July
2022 noted that current FDA guidance does
not clearly explain how bioprinted products
will be regulated, which may cause some
companies to be hesitant about using new
manufacturing technologies like 3D printing.
WHAT’S NEXT?
WHAT IS IT?
BIOPRINTED BONE
REPLACEMENTS AS A
THERAPY FOR ACUTE
BONE INJURIES
VIGNETTE 4
Source: Sutthab/solvod/stock.adobe.com (images). | GAO-23-105430
Regenerative Medicine GAO-23-105430 17
Organoids. Organoids are small, artificially
grown groups of cells or tissues that resemble
an organ and mimic the original tissue
architecture. Organoids can be grown from
patient tissues, and have been successfully
generated from several kinds of human
tissues including heart, liver, brain, and
kidney. Currently, organoids are being used
primarily for research and testing during
multiple stages of the drug development
process. However, researchers are also
evaluating a variety of organoid technologies
to determine whether they may be used to
cure diseases such as diabetes—which affects
how the body uses sugar (see vignette 5).

Source: GAO (analysis); Microone/christosgeorghiou/stock.adobe.com
(images). | GAO-23-105430
A possible process for generating pancreatic organoids.
Type 1 diabetes occurs when a person’s
immune cells attack pancreatic islet cells.
This destroys the person’s ability to produce
insulin, an essential hormone needed to
properly convert sugars to energy and control
blood sugar levels in the human body. About
1.6 million Americans have type 1 diabetes
and need daily insulin injections throughout
their lives, a significant economic burden to
the individual and the U.S. health care system.
Pancreatic islet organoids oer the possibility
of curing the disease by restoring a patient’s
ability to produce insulin.
Pancreatic islet organoids are in phase 1 and
2 clinical trials in humans. Organoid technol-
ogies have significant potential to transform
research and therapeutics. As a research
technology, organoids may model human
disease more accurately than animals and
help drugs move from the laboratory to the
clinic more quickly. As therapeutics, they
may be capable of more complex functions
than simple biological products. However,
it is dicult to predict the future direction
of this technology given its early develop-
ment stage.
WHAT’S NEXT?
WHAT IS IT?
PANCREATIC ISLET
CELL ORGANOIDS AS
A THERAPY FOR TYPE 1
DIABETES
VIGNETTE 5
Source: Rfbsip/solvod/stock.adobe.com (images). | GAO-23-105430
Regenerative Medicine GAO-23-105430 18

Regenerative Medicine GAO-23-105430 19
Full-size organs. Whole organs can be
engineered using the methods described
above. However, full-size engineered organs
for clinical use are still in the early research
and development phase and face several
technical limitations. In order to restore the
function of an organ, all the relevant
components need to be engineered. The
vessels that carry blood and other cells
throughout the body are important, as they
allow oxygen, nutrients, and immune cells to
reach every part of the body. These vessels
are a fundamental feature of most complex
organs, and researchers are studying how to
engineer organs with vascular systems.
Researchers have successfully developed
organs that have less engineering complexity
18
Anthony Atala et al. “Tissue-engineered autologous bladders
for patients needing cystoplasty.” Lancet (London, England),
and used them to cure spina bifida-induced
bladder damage.
18
Lab-grown bladders,
developed from a small piece of a patient’s
bladder, have smooth muscle cells on the
outside and specialized bladder-lining cells on
the inside. Researchers grew both types of
cells separately at first and layered them
together onto a bladder-shaped,
biodegradable scaffold. After further growth,
the bladders were implanted into children
whose spina bifida had damaged the neural
connections that allow nerve cells to help
signal a full bladder (see fig. 4). However, the
use of engineered bladders to treat patients is
currently advancing through clinical trials.
vol. 367, 9518 (2006): 1241-6. https://doi.org/10.1016/S0140-
6736(06)68438-9.
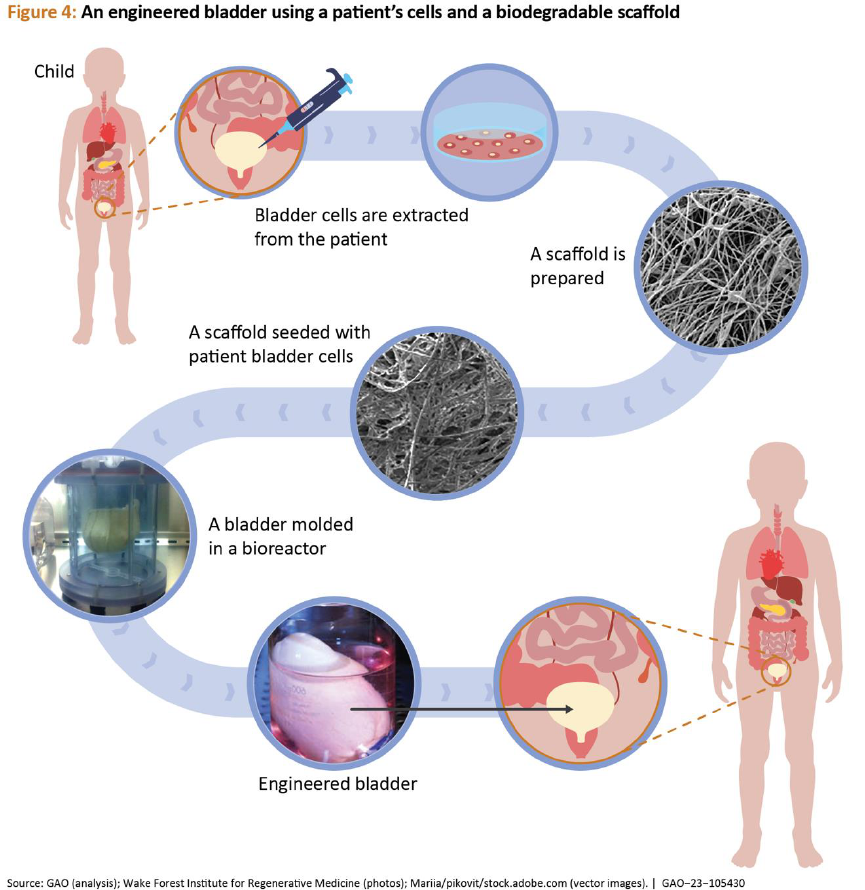
Regenerative Medicine GAO-23-105430 20

Regenerative Medicine GAO-23-105430 21
3 Challenges and Policy Options for Regenerative Medicine
Technologies and Therapies
Regenerative medicine technologies and
therapies have potential benefits, but
challenges may affect their development
and use. We identified challenges including
but not limited to: standardization,
regulations, and manufacturing.
19
GAO developed 11 policy options that could
help address these challenges or enhance the
benefits of regenerative medicine. These
policy options are provided to inform
policymakers of potential actions to address
the policy challenges identified in this
technology assessment. They identify possible
actions by policymakers, which include
Congress, federal agencies, state and local
governments, academic and research
institutions, and industry.
3.1 Challenges related to
standardization
Standardization can help promote more rapid
and effective technology development, but
relatively few standards exist for regenerative
medicine technology. A 2020 FDA-
commissioned report from the Nexight Group
19
We identified other challenges that may affect the
development and use of regenerative medicine technologies
including: Gaps in funding for translational research, market
access and reimbursement, and potential difficulty in
understanding safety of some therapies in the short-term.
20
SCB is a nonprofit organization first established as an
initiative by the Alliance for Regenerative Medicine, but is now
an independent organization that engages industry, academic,
and government stakeholders to accelerate the standards
development process. SCB is also referred to as the Standards
Coordinating Body. See SCB, The Regenerative Medicine
Standards Landscape (Fall 2020),
https://static1.squarespace.com/static/58a331b0db29d63c7fb
and Standards Coordinating Body for Gene,
Cell, and Regenerative Medicines and Cell-
Based Drug Discovery (SCB) identified a strong
need for more standards and outlined more
than 250 needed standards relevant to
regenerative medicine.
20
However,
developing standards is challenging because
these technologies are complex and rapidly
evolving. Developing standards is also
challenging because of the need to reach
consensus across a range of stakeholders and
the need for accurate, well-developed
measurement science in the field.
Standards are rules, conditions, guidelines, or
agreed-upon practices that are adopted
within an industry.
21
They are created to
provide researchers and developers with a
common framework, which promotes
consistency across product development,
manufacturing, and other processes.
Standards are generally developed outside of
the federal government by independent
organizations and are therefore distinct from
federal statutory or regulatory requirements,
unless the regulations are specifically tied to
64528/t/5fc51dfc173fb5383b470452/1606753809117/Landsca
peReportFall2020.pdf, accessed Mar. 3, 2023.
21
Standards include documentary standards, reference
materials, and reference data. Documentary standards are
written documents containing protocols, experimental
methods, technical specifications, or terminologies. Reference
materials are highly characterized substances with known
properties, used to ensure consistency and quality of a
product, calibrate equipment, serve as experimental controls,
or aid in describing and evaluating qualitative and quantitative
data. Reference data are critically evaluated quantitative data
related to a measurable physical or chemical property of a
substance.

Regenerative Medicine GAO-23-105430 22
such standards.
22
For example, the U.S.
Pharmacopeial Convention, a nonprofit
organization, publishes the U.S.
Pharmacopeia: a continuously revised
document that sets quality, purity, and
strength standards for medicines, food
ingredients, and dietary supplements. Small-
molecule drug manufacturers test their
products, which include over-the-counter
drugs like aspirin, against the U.S.
Pharmacopeia’s published standards to help
ensure safety and consistency.
However, regenerative medicine technologies
and therapies are significantly more complex
than small-molecule drugs, in part because
they can be highly personalized and made of
living cells. Currently, regenerative medicine
has relatively few standards, which raised
concerns with some experts we spoke with.
23
For example, a report from a leading
advocacy organization said there is unclear
guidance on how to ensure certain products
22
The National Technology Transfer and Advancement Act of
1995, codified the existing policies in Office of Management
and Budget Circular A-119, “Federal Participation in the
Development and Use of Voluntary Consensus Standards and in
Conformity Assessment Activities.” The act states that the
National Institute of Standards and Technology (NIST) should
facilitate standards-related information sharing and
cooperation between federal agencies and to coordinate the
use by federal agencies of private sector standards
emphasizing where possible, the use of standards developed
by private, consensus organizations. Pub. L. No. 104-113, § 12,
110 Stat. 775, 782 (1996) (codified at 15 U.S.C. § 272(b)(3)).
Similarly, the Office of Management and Budget guidance
states that its policies are intended to encourage federal
agencies to benefit from the expertise of the private sector,
promote federal agency participation in standards bodies to
support the creation of standards that are useable by federal
agencies, and minimize reliance on government-unique
standards where an existing standard would meet the federal
government’s objective. Office of Management and Budget,
OMB Circular No. A-119, Federal Participation in the
Development and Use of Voluntary Consensus Standards and in
Conformity Assessment Activities, (originally issued Oct. 20,
1993, it was subsequently revised and replaced in 1998, and
later revised Jan. 27, 2016).
are sterile, even though such guidance could
significantly reduce the potential for
contamination.
24
SCB also agreed that advancing the
development and use of voluntary consensus
standards in regenerative medicine may
accelerate innovation, increase product safety
and reliability, accelerate regulatory review,
and decrease costs. The 21st Century Cures
Act, enacted in 2016, required the Secretary
of Health and Human Services, in consultation
with the National Institute of Standards and
Technology (NIST) to facilitate an effort to
coordinate and prioritize the development of
standards for regenerative medicine.
25
SCB’s
2020 report stated that a lack of standards
leaves researchers and manufacturers to
independently solve the complex challenges
of clinical translation and scaling of
commercial products. The report also noted
that a lack of standards may raise safety
concerns (see text box) and prevent novel
23
We interviewed experts from government, academia,
industry, and the nonprofit sector, and convened an expert
meeting to discuss the objective topics. See Objectives, Scope,
and Methodology section for more details. The U.S.
Pharmacopeia does not have the authority to create standards
for regenerative medicine. According to the National
Technology Transfer and Advancement Act of 1995 and the
Office of Management and Budget Circular No. A-119, the
federal government prefers the use of standards developed
through a consensus-based process. Standards development
organizations that follow a consensus-based process can be
accredited by the American National Standards Institute and
include organizations like the International Society of
Automation and the International Organization for
Standardization. The U.S. Pharmacopeia does not meet these
requirements and is therefore not recognized as a consensus
standards developing body.
24
Alliance for Regenerative Medicine, A-CELL: A case study-
based approach to integrating QbD principles in Cell-based
Therapy CMC programs, https://alliancerm.org/wp-
content/uploads/2022/09/PROJECT-A-CELL-V2.pdf, accessed
Feb. 22, 2023.
25
Pub. L. No. 114-255, § 3036, 130 Stat. at 1104 (codified at 21
U.S.C. § 356g).

Regenerative Medicine GAO-23-105430 23
regenerative medicine therapies from
becoming commercially viable.
However, overly rigid standards may also
cause problems. FDA officials cautioned that,
at this time, standards for regenerative
medicine should be optional and take a
flexible approach that can account for the
complexity of biological products. They said
that imposing stringent, mandatory
standards, such as those used for small-
molecule drugs, may impede the
development of innovative biologics and
place unnecessary burdens on industry and
on FDA reviewers.
In addition to the complexity of regenerative
medicine technologies and therapies, we
identified the following two challenges that
make it difficult to develop and establish
standards in the field.
Standards require consensus. Standards are
developed through a consensus-building
process that requires participation from a
range of stakeholders. Unlike regulations,
standards can be voluntary and are not
typically developed by government agencies,
so broad buy-in is important for them to be
accepted and used. However, even if
stakeholders agree that a particular standard
should exist, it can be difficult to reach
agreement on the details. This is especially
true if one or more companies have existing
products or infrastructure that do not align
with the proposed standard. For example,
experts noted that companies that have
already built unique data infrastructures are
unlikely to adopt new data standards if
switching would require significant time and
money.
To overcome this barrier and accelerate the
standards development process, SCB engages
with various regenerative medicine
stakeholders in industry, academia, and
government. This engagement has helped
identify, prioritize, and develop voluntary
standards, including standards related to
sterility testing and cell counting. However,
SCB’s impact is limited by its current size and
funding. According to an SCB representative,
SCB receives the majority of its operating
budget through FDA and NIST contracts,
which facilitates federal participation in
standards development but is not sufficient to
address the current need for regenerative
medicine standards. This representative
stated that SCB is hesitant to collect
membership fees because it could limit
stakeholder participation in the standards
development process and would be counter
Standards can help address safety concerns
Viral vectors are commonly used as delivery vehicles for
gene therapy products. The viral vectors insert a modified
DNA sequence into patient’s cells, which can help cure a
wide range of diseases and genetic disorders. However,
according to the Standards Coordinating Body for Gene,
Cell, and Regenerative Medicines and Cell-Based Drug
Discovery (SCB), therapies using viral vectors can produce
adverse and even life-threatening reactions in patients if
administered at the wrong dose.
a
In 1999, a patient died
due to a severe immune response during a gene therapy
trial that used a viral vector. The field lacked a reference
material that could help regulators to adequately evaluate
the safety of such therapies.
In response to this incident, a working group of experts
from industry, academia, and FDA created a standard
reference material for that viral vector: a highly
characterized sample containing a known concentration of
viral vectors. This material, first released in 2002 and used
until 2022, helped developers accurately determine viral
vector concentrations in their products. While developers
are not required to use a reference material, and FDA has
additional processes to establish a product’s safety and
effectiveness, SCB stated that this viral vector reference
material helped address safety concerns and restore
public confidence in gene therapies.
Source: GAO. | GAO-23-105340
a
SCB, Standards Development in Action: Reference Material for Human
Adenovirus 5, https://www.standardscoordinatingbody.org/adenovirus,
accessed Mar. 14, 2023.

Regenerative Medicine GAO-23-105430 24
to the consensus-based process supported by
federal stakeholders.
Two federal agencies—NIST and FDA—have
important roles in standards development.
NIST engages with key stakeholders to
develop consensus and helps ensure that
standards do not conflict with or duplicate
each other.
26
It currently runs laboratory
programs to advance measurements needed
for the characterization and testing of
regenerative medicine manufacturing and
leads multiple consortia to develop or support
the development of documentary standards
and reference materials for regenerative
medicine.
27
Federal law and policy encourage
agencies to use industry-developed standards
whenever possible. NIST therefore works with
appropriate standards development
organizations to advance documentary
standards for regenerative medicine. NIST
also supports the development of reference
materials made available through NIST or
another entity.
FDA also has a role, as FDA officials review
and recognize the voluntary standards that
the agency can apply during its review of
products for regulatory approval. Product
sponsors can choose to follow a voluntary
standard recognized by FDA, which may
reduce the amount of supporting data and
information they need to submit to FDA.
28
However, in response to draft guidance from
26
According to NIST officials, the agency’s role is to support
research and development, translation, and manufacturing,
including characterization and testing, as well as promoting the
broader ecosystem.
27
NIST leads multiple laboratory programs for regenerative
medicine and has a contract with SCB to support standards
development. NIST, RMAT Laboratory Programs,
https://www.nist.gov/regenerative-medicine, accessed Apr. 5,
2023.
FDA, several organizations stated that the
agency’s process for recognizing voluntary
standards has not been clear for regenerative
medicine, and stakeholders may therefore
hesitate to commit resources to developing
standards. FDA published draft guidance on
the Voluntary Consensus Standards
Recognition Program for Regenerative
Medicine Therapies in June 2022, which the
agency said can facilitate the development of
safe and effective regenerative medicine
products.
29
Agency officials told us that
finalized guidance is anticipated to be
published in calendar year 2023.
Additional measurement science is needed.
Measurement science ensures that
measurements are reliable, comparable, and
accurate. Reliable measurements are a key
driver for emerging technologies, but often
require dedicated research that is separate
from technology development. For example,
it took decades of measurement science
research to directly connect the
measurement of time to a fundamental
physical constant—the vibration of a cesium
atom. Once time could be measured
consistently around the globe, new
technologies that rely on highly accurate time
measurements could start to emerge, like
global positioning systems (GPS). Similarly,
advancing measurement science in different
areas of regenerative medicine can support
28
A product sponsor or applicant means any person who
submits or plans to submit an application to FDA for premarket
review. 21 C.F.R. § 3.2(c) (2022).
29
Food and Drug Administration, Voluntary Consensus
Standards Recognition Program for Regenerative Medicine
Therapies (June 16, 2022). Available from:
https://www.fda.gov/media/159237/download, accessed June
16, 2022.
Regenerative Medicine GAO-23-105430 25
standardization and technology development
(see text box).
NIST officials told us that budgetary resources
for regenerative medicine standards, which
includes work on measurement science, have
been limited and inconsistent. Agency officials
also said that fluctuating resources may
hinder efforts to support industry and
advance regenerative medicine standards.
We identified three policy options to help
address challenges in regenerative medicine
standardization. Table 1 presents these
options, along with potential opportunities
and considerations.
Table 1: Policy options for regenerative medicine standardization
Measurement science in regenerative medicine
Sickle cell disease is a genetic condition caused by a one-
letter mutation in the gene for hemoglobin, a protein in
red blood cells. As a result of this genetic mutation, red
blood cells change to a crescent (or sickle) shape and can
cause significant pain. Gene therapies aim to cure sickle
cell disease by changing the incorrect letter without
altering any of the other 3 billion letters in the patient’s
genome. However, it is difficult to measure whether a
gene therapy has created any unintended changes.
DNA sequencing, a measurement technology used to
observe the effects of gene therapies (among other uses),
is imperfect and accuracy can vary depending on the
technique being used. Even the most accurate existing
methods will still take many inaccurate measurements
across a person’s full genome, due to inherent errors in
the process. This creates a critical measurement
challenge, because it will not be clear whether an altered
letter in the data was caused by the gene therapy or the
sequencing method. Improved DNA sequencing
technologies, standards, and reference materials could
therefore increase confidence in gene therapies. Such
improvements will require specific research on the
methods used for sequencing and on
new chemistry or data analysis techniques that could
reduce error.
Source: GAO. | GAO-23-105340
Policy options
Opportunities
Considerations
Invest in standards development
This policy option could help
address the challenge that
standards require consensus.
Potential implementation
approaches:
Government agencies could
support organizations that
develop regenerative medicine
consensus standards.
Government agencies could
support consensus-building
activities between stakeholders,
such as those conducted by the
Standards Coordinating Body for
Gene, Cell, and Regenerative
Medicines and Cell-Based Drug
Discovery.
Could streamline standards
development, which may, in turn,
accelerate innovation, increase
product safety and reliability,
accelerate regulatory review, and
decrease costs of regenerative
medicine therapies.
Existing organizations may not
include all stakeholders, and
stakeholders may hesitate to accept
standards created without their
input.
Industry stakeholders may hesitate
to adopt standards if they perceive
it will cost them a controlling
position in the market.
Standards should be appropriately
flexible to allow for innovation,
while still being detailed and specific
enough to support manufacturing of
consistent, quality products.

Regenerative Medicine GAO-23-105430 26
Source: GAO. | GAO-23-105430
3.2 Challenges related to regulation
According to experts we interviewed, the field
of regenerative medicine faces several
challenges related to regulation, including:
Lack of access to regulatory expertise.
Difficulty navigating a complex regulatory
framework.
Current regulatory pathways may be
insufficient for emerging technologies and
therapies.
Staffing shortages at FDA and
collaborating agencies.
Unlicensed stem cell products.
Lack of access to regulatory expertise.
Sponsors who develop regenerative medicine
products need regulatory expertise
throughout all stages of product
development, including the stage where they
submit a product for FDA review. Start-ups
and other small companies or academic
institutes that do not have designated in-
house regulatory departments may be at a
disadvantage due to lack of expertise on the
Provide more consistent support
for measurement science
research.
This policy option could help
address the need for more
measurement science.
Potential implementation
approaches:
Government agencies (e.g., NIST,
FDA) could dedicate specific
funding for measurement science
research.
Industry stakeholders could
devote more resources to
measurement science research
initiatives.
Could enable more or faster
development of regenerative
medicine technologies, and could
provide additional benefits
outside of regenerative medicine.
Additional federal spending on
measurement science for
regenerative medicine may shift
resources that were supporting
other emerging technologies.
Private industry may not invest in
measurement science since they
may not receive a timely return on
investment.
Maintain the status quo.
Standards may be developed
without further intervention.
Potential cost savings for federal
agencies.
Would avoid establishing
standards too early, which can
stifle innovation and competition.
Technologies may be more likely to
fail in development or during
regulatory review due to a lack of
standardization.
Companies may need to spend more
to meet safety requirements for
FDA approval or licensure.
Companies may not be willing to
change their existing processes if
standards are not established early
enough.

Regenerative Medicine GAO-23-105430 27
complex regulatory process. This lack of
expertise could delay the product
development process. For example,
companies could spend time and resources
generating data that do not meet FDA
requirements. An expert told us that these
companies need access to knowledgeable
regulatory experts and adequate
opportunities to interact with FDA
reviewers.
30
Difficulty navigating a complex regulatory
framework. Clear and predictable regulations
ensure that product developers are able to
understand the data and other requirements
needed for approval without unnecessary
delays or uncertainty. Experts told us that it
can be challenging for product sponsors to
navigate the complex regulatory framework
for regenerative medicine products, which
may span multiple FDA centers and pathways
to approval. Some regenerative medicine
products are combination products (see sec.
2.2), and it can be difficult to understand
what classification they fall under.
31
While
some regenerative medicine products clearly
fit in to a particular classification, others may
be less clear. This can be challenging for
technologies and therapies for which the
primary mode of action may not be known or
fully understood.
Another layer of complexity comes from the
multiple FDA programs for which
30
In order to address the substantial growth in the
development of novel products, CBER has established a new
Office of Therapeutic Products. This reorganization is intended
to create flexibility and capacity for future growth in the
number of full-time employee positions and enhance the
timeliness and consistency of the office’s interactions with
sponsors.
31
Officials told us that the Office of Combination Products
situated in FDA’s Office of the Commissioner evaluates the
classification and regulatory review jurisdiction of combination
regenerative medicine products may be
eligible. Sponsors of regenerative medicine
products can ask FDA to review their product
under one or more of these programs if they
meet the criteria. For example, they can
request RMAT designation, which allows for
accelerated approval of products with the
potential to address unmet medical needs. In
addition, regenerative medicine products may
be eligible for other expedited programs,
including fast track designation, breakthrough
therapy designation, accelerated approval,
and priority review designation.
Sponsors may
receive more than one designation for a given
product, but they must request each one
separately.
32
To understand which programs are available
before submitting, product sponsors can get
information in many ways including seeking
advice from FDA. Experts told us that FDA was
generally inclined to provide advice to
sponsors who ask for it, but the agency does
not always have the ability to respond as
quickly as it would like. We also heard from
experts that such advice, when provided to
regenerative medicine product sponsors, is
not always clear, leading sponsors to spend
extra time seeking information. Experts also
told us that sponsors could benefit from clear
guidance documents and additional
opportunity for interaction with FDA
reviewers at various stages of product
products as well as other articles for which the classification as
drug, device, and/or biologic is unclear. Officials told us that
the Office of Combination Products has various mechanisms for
stakeholders to obtain recommendations or determinations for
their products.
32
Priority review designation is determined for every product
application, regardless of whether the product sponsor
requested it.

Regenerative Medicine GAO-23-105430 28
development.
33
They said clarifying guidance
to sponsors as early as possible in the product
development process could save time and
resources, potentially making therapies
available to patients sooner.
34
Current regulatory pathways may be
insufficient for emerging technologies.
Emerging regenerative medicine technologies
and therapies may blur the lines between
drugs, biologics, and devices which could
make their pathways to approval or licensure
more uncertain. Participants in our expert
meeting and other experts told us that the
requirements for these types of products can
unintentionally hinder the development of
emerging technologies. To illustrate this,
experts told us about medical devices made
from materials that can promote cell growth
or tissue healing. One such device is known as
a tissue fixation implantable device which is
used to attach soft tissue grafts to a fractured
bone to promote healing. These experts told
us that products made with such materials
are regulated as devices, and FDA guidance
does not allow sponsors to claim regenerative
properties for products regulated solely as
devices.
35
However, experts said there is a
growing understanding that devices may be
more effective if they are made from
33
Sponsors can obtain early feedback from FDA through an
Initial Targeted Engagement for Regulatory Advice on
CBER/CDER Products (INTERACT), which is a meeting at a
specific time early in product development.
34
GAO previously reported similar challenges with existing FDA
guidance related to advanced manufacturing for drugs. See
GAO, Drug Manufacturing: FDA Should Fully Assess Its Efforts
to Encourage Innovation, GAO-23-105650 (Washington, D.C.:
Mar. 10, 2023).
35
According to FDA, combination products can be eligible for
RMAT designation when the biologic constituent part of the
product is a regenerative medicine therapy and that therapy
serves as the product’s primary mode of action.
materials that are biologically active and
promote cell regeneration.
Another possible difficulty is that companies
that develop novel products may lack
examples of the same types of products
previously going through the regulatory
process. Experts said that this can lead to
confusion about which regulatory pathway is
most appropriate for their product.
36
We
heard from experts that there is an
opportunity for FDA to clarify regulatory
pathways for regenerative medicine products
and assess the need for alternative pathways.
For example, experts said that a new pathway
or amendments to current pathways could be
proposed to allow for devices with
regenerative properties.
Staffing shortages at FDA and collaborating
agencies. FDA needs knowledgeable
personnel to handle incoming applications,
provide clear advice to product sponsors, and
achieve the agency’s mission of advancing
public health. Agencies that collaborate with
FDA and fund regenerative medicine
programs, like the National Institutes of
Health, also need to have personnel
36
GAO previously reported that FDA is working to clarify and
address similar challenges related to the agency’s review of
drugs made using advanced manufacturing technology. For
example, FDA has a website that has a list of technologies that
have been accepted into CDER’s Emerging Technology
Program, thus informing industry stakeholders about the type
of technologies FDA has experience reviewing. In addition,
CDER is implementing an initiative to examine its regulatory
framework for advanced manufacturing to determine whether
changes are needed to its statutory authorities, regulations,
and guidance in order to facilitate the agency’s review of
applications that use advanced manufacturing technologies.
See GAO-23-105650.

Regenerative Medicine GAO-23-105430 29
knowledgeable in regulatory science.
37
We
heard from experts that even when provided
with a potentially sufficient number of
positions, agencies have historically faced
challenges meeting their medical product
workforce needs, due in part to competition
with the private sector. Experts we spoke
with said that FDA continues to lack adequate
capabilities, including the ability to recruit,
train, and retain regulatory scientists.
Without sufficient interdisciplinary training,
FDA reviewers may be less familiar with novel
and complex emerging regenerative medicine
technologies.
38
Experts told us that this can
lead to inconsistent or contradictory advice
over the course of product development.
Experts conveyed the importance of
bolstering FDA’s ability to hire and retain
reviewers trained in evaluating emerging
technologies and therapies. A recent GAO
report recommended that FDA develop and
implement an agency-wide strategic
workforce plan with performance measures
to ensure it can evaluate the effectiveness of
its human capital efforts.
39
Unlicensed stem cell products. Stem cells can
be the basis for safe and effective treatments
and FDA has licensed stem-cell products
37
Regulatory science is the science of developing new tools,
standards, and approaches to assess the safety, efficacy,
quality, and performance of all FDA-regulated products.
https://www.fda.gov/science-research/science-and-research-
special-topics/advancing-regulatory-science, accessed July 6,
2023.
38
FDA hosts the Centers of Excellence in Regulatory Science
and Innovation (CERSI) program to foster robust and innovative
approaches to advance regulatory science, and the goal is for
the CERSIs to advance regulatory science individually and
synergistically through collaborative interactions with FDA
scientific experts and funding offices.
39
GAO, FDA Workforce: Agency-Wide Workforce Planning
Needed to Ensure Medical Product Staff Meet Current and
derived from cord blood for limited use in
patients with blood disorders. However, some
U.S. clinics offer stem cell products that are
not FDA licensed. Experts told us that such
clinics are eroding public trust in regenerative
medicine technologies and therapies, and are
a threat to public health and safety. According
to a study in 2021, more than 2,700 clinics
were found selling purported stem cell
treatments in the U.S.
40
In 2019, FDA issued a
warning about stem cell treatments that are
illegal and potentially harmful and asked
patients to ensure any treatments they are
considering are either FDA licensed or part of
an FDA-approved study.
41
FDA stated that it
“is increasing its oversight and enforcement
to protect people from dishonest and
unscrupulous stem cell clinics, while
continuing to encourage innovation so that
the medical industry can properly harness the
potential of stem cell products.”
Other government agencies and states are
also taking action against clinics marketing
certain unlicensed stem cell products. For
example, in 2021, the Federal Trade
Commission (FTC) and the Georgia Attorney
General’s Office sued the co-founders of the
Stem Cell Institute of America for allegedly
Future Needs, GAO-22-104791 (Washington, D.C.: Jan. 14,
2022).
In response, FDA stated in July 2022 that it was working to
develop and implement an agency-wide strategic workforce
plan to document human capital goals, and anticipates having a
baseline version of this plan by the end of fiscal year 2024. GAO
will continue to follow the agency's progress on this activity.
40
Turner, Leigh. “The American stem cell sell in 2021: U.S.
businesses selling unlicensed and unproven stem cell
interventions.” Cell stem cell vol. 28, 11 (2021): 1891-1895.
https://doi.org/10.1016/j.stem.2021.10.008.
41
Food and Drug Administration, FDA Warns About Stem Cell
Therapies (Washington, D.C.: Sept. 3, 2019).
https://www.fda.gov/consumers/consumer-updates/fda-
warns-about-stem-cell-therapies, accessed Mar. 28, 2023.

Regenerative Medicine GAO-23-105430 30
marketing stem cell therapy to seniors
nationwide using “bogus claims” that it is
effective in treating arthritis, joint pain, and a
range of other orthopedic ailments.
42
FTC also
issued a warning about false and misleading
information about stem cell therapies, as a
number of them have not been shown to be
safe or effective.
43
A recent report suggests that patients
considering stem cell and regenerative
medicine interventions do research online or
by contacting friends, family, medical
providers, and consultation services.
44
However, a 2021 study concluded that efforts
should be directed at helping physicians
42
Federal Trade Commission et al v. Peyroux et al, 1:21-vc-
03329 (N.D. Ga. Filed Aug. 16, 2021).
43
FTC, Think Stem Cell Therapy Can Treat Your Ailments? It
may pay to think twice (Aug. 17, 2021),
https://consumer.ftc.gov/consumer-alerts/2021/08/think-
stem-cell-therapy-can-treat-your-ailments-it-may-pay-think-
twice, accessed on Mar. 26, 2023.
44
Arthurs, Jennifer et al, “Patients seeking stem cell
therapies—a prospective qualitative analysis from a
obtain information to inform themselves and
their patients about unlicensed regenerative
medicine therapies.
45
Experts warned that the
public may be vulnerable to confusion and
the spread of false information online,
partially because of the novelty and
complexity of these emerging technologies.
We identified five policy options to help
address challenges related to the regulation
of regenerative medicine products. Table 2
presents these options, along with the option
of maintaining the status quo, and
opportunities and considerations.
Regenerative Medicine Consult Service,” npj Regenerative
Medicine (2022) 7:20. https://doi.org/10.1038/s41536-022-
00215-w.
45
Smith, Cambray et al, “Academic Physician Specialists’
Approaches to Counseling Patients Interested in Unproven
Stem Cell and Regenerative Therapies - A Qualitative Analysis,”
Mayo Clinic Proceedings, vol. 96, 12 (2021): 3086-3096.
https://doi.org/10.1016/j.mayocp.2021.06.026.

Regenerative Medicine GAO-23-105430 31
Table 2: Policy options for regenerative medicine regulation
Policy options
Opportunities
Considerations
Provide opportunities for
increased interactions between
regulatory experts (at FDA or in
industry) and smaller companies,
especially early in the
development process.
This policy option could help
address the lack of access to
regulatory expertise.
Potential implementation
approaches:
Policymakers could increase
funding to existing public-private
partnerships that can provide
access to regulatory experts.
Sponsors could devote more
resources to sharing lessons
learned from their regulatory
submissions to help accelerate
technology development across
the field.
May provide more timely advice
and avoid unnecessary delays or
uncertainty by pursuing the
wrong regulatory pathways or
submitting data that do not meet
regulatory requirements.
May require additional resources
to bolster the workforce of
regulatory scientists at FDA or
public-private partnerships.
FDA may be limited in its ability to
advise companies early in the
process so as not to create a
conflict of interest.
Identify mechanisms for FDA to
clearly communicate advice for
regenerative medicine product
classification and update
guidance documents
accordingly.
This policy option could help
address the challenge of
navigating a complex regulatory
framework.
Potential implementation
approaches:
FDA could provide examples in
guidance documents to further
clarify product classifications.
Examples could be provided for
technologies and therapies that
FDA has experience reviewing.
FDA could provide mechanisms
to ensure consistent advice
across FDA reviewers when
Could encourage new products
and may speed up the review
process.
Examples can further clarify
product classifications.
The rapidly changing field of
regenerative medicine may
necessitate more frequent
updates to guidance documents.
Guidance that is too specific can
be a constraint if there are
multiple valid ways of doing
things.

Regenerative Medicine GAO-23-105430 32
Policy options
Opportunities
Considerations
responding to product sponsor
inquiries.
Consider whether changes to the
framework for evaluating
combination products and
medical devices to
accommodate emerging
technologies and therapies may
be necessary.
This policy option could help
address whether current
regulatory pathways are
sufficient for emerging
technologies and therapies.
Potential implementation
approaches:
FDA could consult with other
stakeholders to determine
whether amendments to existing
pathways or additional
pathways are needed.
The framework could allow
products regulated solely as
medical devices and made from
materials that promote cell
growth or tissue healing to claim
regenerative properties.
May encourage innovators,
researchers, and developers of
new products to provide
valuable feedback to regulators.
Coordinating among stakeholders
to consider changes to regulatory
pathways may be time- and
resource-intensive.
If such consideration leads to
recommended changes to the
framework, statutory and
regulatory changes may be
necessary.
Improve FDA’s ability to develop
and maintain an appropriate
interdisciplinary regulatory
workforce.
This policy option could help
address the challenge of staffing
shortages at agencies like FDA.
Potential implementation
approaches:
FDA could continue to develop
and implement an agency-wide
strategic workforce plan.
FDA could improve training for
current staff on the latest
technologies and therapies.
Could result in timely feedback
to sponsors, enable increased
interaction between reviewers
and sponsors, and make
therapies available to patients
sooner.
Could require funding for FDA for
additional positions.
Salaries may need to be increased
for FDA to compete with the
private sector.
Support better and more
effective information tools that
Combat false information and
improve public trust.
Help patients to evaluate the
legitimacy of available therapies.
A public education campaign could
require significant resources, and
it is unclear how its effectiveness
would be evaluated.

Regenerative Medicine GAO-23-105430 33
Policy options
Opportunities
Considerations
are publicly available to
clinicians and patients.
This policy option could help
address the challenge of
unlicensed stem cell products.
Potential implementation
approaches:
Key stakeholders—such as
councils or associations of
governments or federal or state
agencies—could coordinate
strategic campaigns and
partnerships between
government health agencies and
organizations that have broad
public appeal (e.g., faith-or
community-based organizations,
sports, or patient advocacy
groups).
FDA and state health
departments or medical boards
could create and publicize a
shared database of clinics
offering unlicensed stem cell
products.
Federal agencies and
organizations that help
consumers gauge the value,
quality, or authenticity of goods
and services could create
informational materials with
strategies for consumers to
evaluate medical claims and
advertising.
Help increase the diversity of
clinical trial participants, which
improves understanding of the
safety and effectiveness of
medical products for different
populations.
Even with more accurate
information, patients ultimately
decide what is best for their
health based on their personal
circumstances. For example,
studies show that patient
decisions on whether to undergo
an unapproved or unlicensed
intervention are complex and
depend on the patient’s condition,
consideration of medical risks,
trust in research or medical
institutions, and other factors.
Maintain the status quo.
• Could allow current regulatory
framework for evaluating
regenerative products to remain
unchanged.
• Could save government or
private sector resources for
other priorities, including
promising medical technologies
and therapies other than
regenerative medicine.
• Would avoid making changes to
regulatory framework that may
not address the needs of
technologies and therapies yet
to be developed.
• Product developers may have
difficulties advancing new
technologies and therapies to the
market.
• Larger companies may continue to
maintain advantage such as access
to regulatory advisors over smaller
companies.
• Consumers may continue to fall
prey to misleading marketing
about unapproved or unlicensed
stem cell products.
Source: GAO. | GAO-23-105430

Regenerative Medicine GAO-23-105430 34
3.3 Challenges related to
manufacturing
Manufacturing is the creation of new
products from starting materials, in a way
that is generally consistent and
reproducible.
46
It is a key step for many
emerging technologies and therapies,
because it can help increase product
consistency, decrease costs, and facilitate
larger production volumes that make
products more accessible and affordable.
Biologics, including cells, tissues, and organs
used for regenerative medicine are difficult to
manufacture at scale because they are far
more complex than many other medical
products, such as small-molecule drugs (see
text box). This complexity also contributes to
three challenges related to manufacturing in
regenerative medicine: lack of infrastructure,
ensuring quality, and workforce shortages.
Currently, some components of certain
regenerative medicine products can be
reliably manufactured. For example, the DNA
and viral vectors used to alter a cell’s
genome—a key part of gene and cell
therapies—can be produced at large scales.
However, producing complete products, such
as CAR T cell that may cure certain types of
cancer, currently requires technicians to
perform many steps manually. Experts stated
that regenerative medicine technologies and
therapies will require increased levels of
automation if they are to be widely accessible
and affordable.
46
Starting or ancillary materials are materials used during the
manufacturing of cell and tissue products that are not intended
to be a part of the therapy itself.
We identified the following three challenges
to the widespread and efficient manufacture
of regenerative medicine products.
Lack of infrastructure. The cell, tissue, and
organ products being developed for
regenerative medicine will require more
complex manufacturing facilities than are
currently used to produce small-molecule
drugs. For example, many existing
pharmaceutical manufacturing lines are not
entirely closed off from the external
environment, because small-molecule drugs
can be sterilized once manufacturing is
complete, using tools like heat, chemicals,
and radiation. Regenerative medicine
products cannot be sterilized, because
sterilization can damage or kill cells and
tissues. Therefore, manufacturing facilities
will need complex systems to prevent
Biologics are significantly more complex than small-
molecule drugs
Even though small-molecule drugs and biologics are often
discussed in similar contexts, their complexity differs
substantially. Aspirin is a drug that has 21 atoms.
Monoclonal antibodies, which experts consider to be
relatively simple biologics, have around 25,000 atoms. Thus,
the difference in complexity between small-molecule drugs
and monoclonal antibodies is similar to the difference
between a bicycle and a commercial jet.
A human cell is far more complex than a single protein, like
an antibody. An average cell is estimated to contain 42
million proteins, and the precise composition of these
proteins is constantly changing as the cell uses energy and
grows.
a
Existing manufacturing technologies, even those used to
manufacture simpler biologics, require significant
adaptations and advances to manufacture the complex
biologics needed for regenerative medicine.
Source: GAO analysis. | GAO-23-105430
a
Brandon Ho et al. “Unification of Protein Abundance Datasets Yields
a Quantitative Saccharomyces cerevisiae Proteome” Cell Systems
vol. 6, 192–205 (2018). https://doi.org/10.1016/j.cels.2017.12.004.

Regenerative Medicine GAO-23-105430 35
contamination and keep products sterile
throughout manufacturing. Additionally,
manufacturing facilities will need to allow for
some customization to individual patients,
while also enabling some level of mass
production to reduce costs.
Experts stated that standing up such facilities
will be risky for private companies. The
necessary complexity will require significant
investment, regardless of whether the
facilities are newly built or remodeled.
Further, a company likely will not receive a
return on this investment until FDA has
licensed its product, a process that generally
takes years and is difficult to predict early in
product development, according to experts.
Some initiatives are underway to help
companies develop their manufacturing
processes at testbed facilities before building
at larger scales. These facilities, sometimes
operated as public-private partnerships, can
help smaller companies pilot their
manufacturing processes or begin scaling up
production, before they engage with larger
companies or contract manufacturers. For
example, the Advanced Regenerative
Manufacturing Institute (ARMI) has a shared
47
The Departments of Commerce, Defense, and Energy have
established a network of innovation institutes—known as
Manufacturing USA institutes—to promote research,
development, and commercialization of advanced
manufacturing technologies. ARMI is a non-profit organization
administering BioFabUSA, a Manufacturing USA institute (also
known as a Manufacturing Innovation Institute) founded in
2017 and funded by the Department of Defense. Its goal is to
make practical the scalable, consistent, and cost-effective
manufacturing of cells, tissues, and organs. The National
Institute for Innovation in Manufacturing Biopharmaceuticals is
another manufacturing innovation institute funded by NIST,
whose mission is to accelerate biopharmaceutical innovation,
including in the area of cell therapies. GAO is mandated to
facility where member organizations can test
and develop manufacturing processes for new
products.
47
Similarly, the Wake Forest
Institute for Regenerative Medicine has a
manufacturing facility that helps researchers
test manufacturing processes as they develop
their technologies. Additionally, the California
Institute for Regenerative Medicine is
planning to build a California Cell and Gene
Therapy Manufacturing Network that will
address manufacturing bottlenecks and help
advance regenerative medicine therapies to
patients. However, industry experts, including
those at ARMI, stated that more facilities may
be needed to meet the demands of the
regenerative medicine industry. In particular,
patients receiving therapies that use their
own cells or tissues could benefit from
distributed manufacturing facilities to help
increase production capacity and allow
patients to receive therapies more quickly.
48
A
greater and more widely distributed number
of manufacturing facilities may be beneficial,
because there are few existing facilities and
patient cells must be flown to one of those
facilities from hospitals around the country
(see fig. 5).
regularly assess the operation of this network. See, for
example, GAO, Advanced Manufacturing: Innovation Institutes
Report Technology Progress and Members Report Satisfaction
with Their Involvement, GAO-22-103979 (Washington, D.C.:
Dec. 16, 2021).
48
Distributed manufacturing is a decentralized manufacturing
strategy in which portable manufacturing units may be
deployed to multiple locations. Point-of-care manufacturing is
a type of distributed manufacturing in which manufacturing
units are deployed to places close to where patients may
receive care, such as a health care facility. Point-of-care
manufacturing could thus be used by health care facilities to
meet specific patient needs.
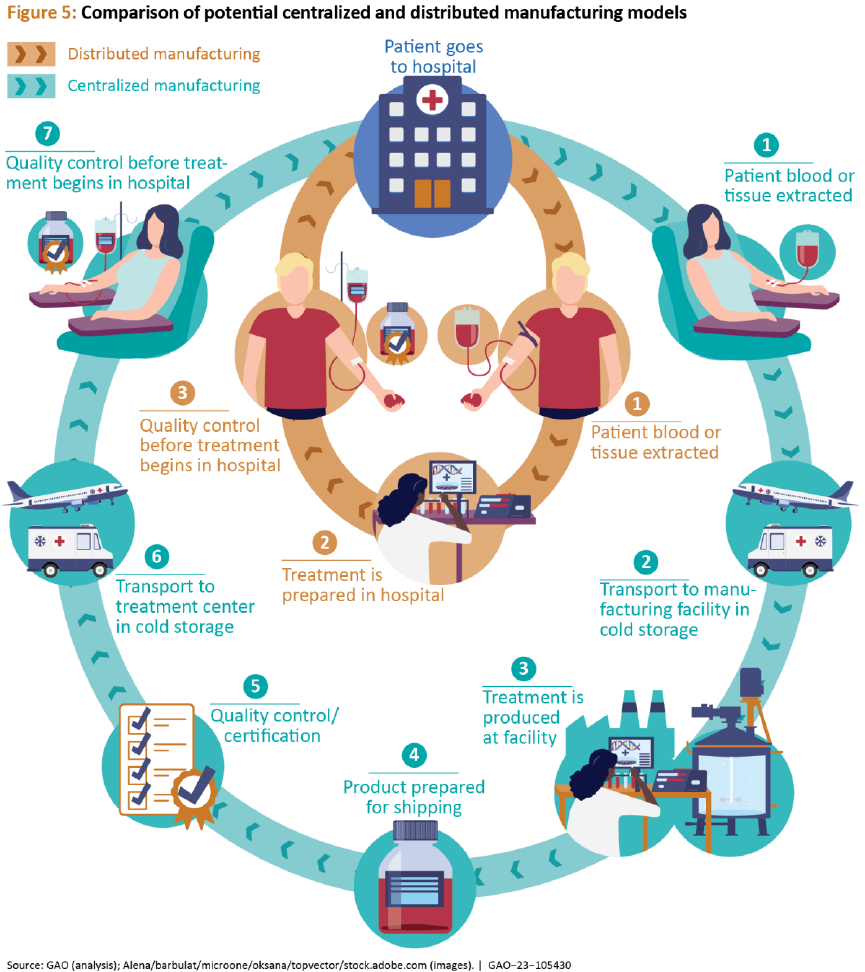
Regenerative Medicine GAO-23-105430 36
Ensuring quality. All medical products have
defined properties or characteristics that help
ensure quality, known as critical quality
attributes (CQA). For example, the CQAs for a
small-molecule drug like aspirin might be the
active ingredient’s concentration or the
product’s purity. These properties can be
measured by, for example, comparing them

Regenerative Medicine GAO-23-105430 37
to established standards.
49
A batch of a drug
can be stored as a reference so future batches
can be compared against it.
However, stakeholders often lack consensus
on how to measure quality for regenerative
medicine products. There are also few
standardized reference materials that can be
used to evaluate a finished product, making it
difficult to identify CQAs. Furthermore,
because regenerative medicine products
contain living cells, they can change over time
or with environmental conditions. For
example, cells in a laboratory may function
differently than the same cells in a patient.
Instead of comparing their products to
reference materials, many regenerative
medicine manufacturers operate under the
assumption that if their manufacturing
processes are consistent, the final product
will be high quality and consistent. FDA has
issued several guidance documents to help
product developers identify CQAs.
50
However,
CQAs are often product-specific and may be
challenging to identify during early clinical
development. Therefore, additional studies
may be needed later in development to
49
Standards for manufacturing may include reference
materials, or internal standards that a company establishes for
their specific products and processes.
50
Food and Drug Administration, Human Gene Therapy
Products Incorporating Human Genome Editing (Mar. 2022),
https://www.fda.gov/media/156894/download, accessed Apr.
11, 2023; Considerations for the Development of Chimeric
Antigen Receptor (CAR) T Cell Products (Mar. 2022),
https://www.fda.gov/media/156896/download, accessed Mar.
8, 2023; Chemistry, Manufacturing, and Control (CMC)
Information for Human Gene Therapy Investigational New Drug
Applications (INDs) (Jan. 2020),
https://www.fda.gov/media/113760/download, accessed Mar.
8, 2023; and Potency Tests for Cellular and Gene Therapy
Products (Jan. 2011),
https://www.fda.gov/media/79856/download, accessed Apr.
11, 2023.
51
Recent FDA draft guidance acknowledges that the safety and
quality of starting and ancillary materials can vary widely
update CQAs for each regenerative medicine
product and establish processes to measure
them.
Keeping manufacturing consistent may also
be difficult because the starting materials
used in regenerative medicine are inherently
variable in their composition. For example, in
the area of stem cell therapies, starting
materials include nutrients for growing cells
and growth factors for triggering stem cells to
grow into the specific type of cell needed for
a therapy. Variation in these materials can
reduce product consistency or cause
contamination.
51
Product quality standards
and oversight may reduce such variation, but
according to experts we spoke with, few
quality standards currently exist for these
materials or even for the starting materials
used to make them.
52
Workforce shortages. According to industry
experts, there is a shortage of skilled technical
personnel who could work on regenerative
medicine manufacturing lines. As demand for
regenerative medicine products grows,
workforce needs will also continue to grow.
depending on factors such as source or vendors. They also note
that lot-to-lot variability and stability of reagents can be
problematic. Food and Drug Administration, Considerations for
the Development of Chimeric Antigen Receptor (CAR) T Cell
Products. (Mar. 2022).
https://www.fda.gov/media/156896/download, 9, accessed
Mar. 13, 2023. Experts told us that, unlike sponsors, starting
material suppliers are not required to follow current good
manufacturing practice regulations if a material is not
incorporated into a final product.
52
The International Organization for Standardization has
recently published a standard that gives guidance to suppliers
and users of ancillary materials to improve the consistency and
quality of ancillary materials used in the production of cellular
therapeutic products and gene therapy products for human use.
International Organization for Standardization, “ISO 20399:2022
Biotechnology – Ancillary materials present during the production
of cellular therapeutic products and gene therapy products,”
December 2022. https://www.iso.org/standard/79399.html,
accessed Mar. 13, 2023.

Regenerative Medicine GAO-23-105430 38
Additionally, manufacturing may need to be
spread out geographically because some
regenerative medicine products would be
easier to produce near patient care centers.
The industry may therefore need technical
workers in many locations, not just cities that
already have a large biomedical workforce.
A recent study noted that regenerative
medicine manufacturing requires people to
perform routine, repetitive processes with as
much consistency as possible.
53
Experts also
suggested that workers will need technical
skills, such as the ability to accurately handle
liquids and keep materials sterile, but they
may not need significant theoretical
background in biology. Experts said
community and technical colleges may be
best suited to train students for such careers,
because they have robust workforce
development programs. The National Science
53
Gary M. Green et al, “Recommendations for workforce
development in regenerative medicine biomanufacturing,”
Stem Cells Translational Medicine, (2021) 10; 1365-1371.
https://doi.org/10.1002/sctm.21-0037. FDA ensures the quality
of drugs and biologics by monitoring manufacturers’
compliance with its current good manufacturing practice
regulations. 21 C.F.R. Parts 210, 211, 212, and 600 (2022).
54
For more information about the National Science Foundation
Advanced Technological Education (ATE), see
Foundation Advanced Technical Education
program is supporting some workforce
development programs for biotechnology
training, but an expert emphasized the need
to expand to more campuses and increase
awareness about regenerative medicine at
the pre-college level.
54
We previously
reported on the regenerative medicine
workforce and found that, in addition to a
shortage of existing skilled laboratory and
manufacturing technicians, vocational and
technical education is insufficient to meet
both current and future workforce needs.
55
We identified three policy options to help
address these manufacturing challenges.
Table 3 presents these options, along with the
option of maintaining the status quo and
opportunities and considerations.
https://beta.nsf.gov/funding/opportunities/advanced-
technological-education-ate, accessed Feb. 21, 2023.
55
GAO, Regenerative Medicine and Advanced Therapies:
Information on Workforce and Education, GAO-23-106030
(Washington, D.C.: Mar. 23, 2023). We also found that there
were no nationally recognized regenerative medicine
education curricula for various postsecondary degrees.

Regenerative Medicine GAO-23-105430 39
Table 3: Policy options for regenerative medicine manufacturing
Policy options
Opportunities
Considerations
Create more shared pilot- and
mid-scale manufacturing
facilities to help companies
develop their manufacturing
processes.
This policy option could help
address the lack of
manufacturing infrastructure.
Potential implementation
approaches:
Government agencies could
support more public-private
partnerships that can share costs
for manufacturing facilities.
Industry stakeholders could
partner with academic
researchers to increase
manufacturing readiness of
technologies and prepare them
for commercialization.
May accelerate product
development.
May help companies de-risk their
products by giving them
opportunities to develop and
confirm the effectiveness of
automated and scalable
manufacturing processes.
May save time and money by
allowing companies to postpone
building infrastructure until after
their products and manufacturing
processes are further along the
development pipeline.
It will be costly to build shared
manufacturing infrastructure.
It is unclear which stakeholders
should be responsible for funding
and operating shared facilities.
Not all therapies require the same
level of scale-up (e.g., therapies for
rare diseases have smaller market
sizes, so fewer doses will be
needed).
Not all stakeholders agree that
there should be a federal role and
may, instead, prefer to maintain the
current free-market model for
developing regenerative medicine
products.
Issues may arise when sponsors
transition from development
processes in one location to
commercial processes in a second
location.
Proprietary manufacturing
processes may be a component of
FDA licensure. If FDA were engaged
with private companies in
developing such processes, FDA
would need to ensure there was no
conflict of interest and that other
companies had a level playing field.
Provide more oversight and
feedback to suppliers to increase
consistency in starting materials.
This policy option could help
address inconsistency in starting
materials for manufacturing.
Potential implementation
approaches:
FDA could work with the
Standards Coordinating Body for
Gene, Cell, and Regenerative
Medicines and Cell-Based Drug
Discovery and manufacturers to
establish quality standards for
starting materials.
May accelerate manufacturing by
reducing variation in input
materials.
May reduce the risk of failure
during product development.
Starting material suppliers may lack
incentives to follow standards if
they lead to higher costs.

Regenerative Medicine GAO-23-105430 40
Source: GAO. | GAO-23-105430
Starting material suppliers could
commit to following starting
material consensus standards,
like those published by the
International Organization for
Standardization.
Create hands-on training
programs at community and
technical colleges to address
workforce shortages.
This policy option could help
address manufacturing workforce
shortages.
Potential implementation
approaches:
Academic stakeholders could use
government-run pilot facilities to
train students.
Academic stakeholders could
create standardized training
certifications to expand
opportunities for both trainees
and employers.
Could expand the regenerative
medicine workforce and help
students develop technical skills
to meet existing and future needs.
Could lead to increased domestic
manufacturing, which can
contribute to U.S. global
competitiveness.
Could create opportunities for
high-paying jobs that do not
require an advanced degree.
Educational programs may need to
be integrated with regenerative
medicine research programs to
ensure that trainees can stay up to
date on techniques and
technologies.
Community and technical colleges
may have limited access to training
facilities.
Maintain the status quo.
Could allow manufacturing to
continue its current incremental
development.
Could save government or private
sector resources for other
priorities, including promising
medical technologies other than
regenerative medicine.
Larger companies may continue to
control manufacturing.
Product developers may have
difficulty accessing manufacturing
facilities during development,
creating high potential for product
failure once they begin
manufacturing at scale.
Regenerative Medicine GAO-23-105430 41
4 Agency and Expert Comments
We provided a draft of this product to Department of Health and Human Services’ FDA and
National Institutes of Health, Department of Defense, and Department of Commerce’s NIST for
review. Department of Defense concurred without comment. The other agencies and some
participants from our expert meeting provided technical comments, which we incorporated as
appropriate.
We are sending copies of this report to the appropriate congressional committees and other
interested parties. In addition, the report is available at no charge on the GAO website at
https://www.gao.gov.
If you or your staff have any questions about this report, please contact me at (202) 512-6888 or
[email protected]. Contact points for our Offices of Congressional Relations and Public Affairs
may be found on the last page of this report. GAO staff who made key contributions to this
report are listed in appendix III.
Karen L. Howard, PhD
Acting Chief Scientist and Director,
Science, Technology Assessment, and Analytics
Regenerative Medicine GAO-23-105430 42
Appendix I: Objectives, Scope,
and Methodology
Objectives
This report examines:
(1) current and emerging regenerative
medicine technologies and therapies and
their potential benefits,
(2) challenges that hinder the development
and use of regenerative medicine
technologies and therapies, and
(3) policy options that could help enhance
benefits and mitigate challenges associated
with these technologies and therapies.
Scope and methodology
To address all three of our objectives, we
assessed available and developing
regenerative medicine technologies and
approaches that may restore cell, tissue, and
organ functions lost to disease or injury. For
all of our objectives, we reviewed peer-
reviewed scientific literature and other
documents describing current and developing
technologies; interviewed federal agency
officials and experts from government,
academia, industry, the nonprofit sector, and
end user groups such as patient groups; and
convened a 3-day expert meeting with
assistance from the National Academies of
Sciences, Engineering, and Medicine (National
Academies) to discuss the objective topics.
We provide more details on these
methodologies below. We also reviewed
federal agency guidance on the development
and deployment of relevant technologies,
such as Food and Drug Administration
(FDA) guidance on the biologics license
applications process.
Limitations to scope
The list of key technologies discussed in this
report is not intended to be exhaustive. Based
on our review of the literature and
discussions with federal agency officials and
other experts, we selected technologies
currently in use or under development by
researchers to restore body functions that
may be lost to disease or injury. We did not
include technologies used for research
purposes, testing, or diagnostics, such as
organ-on-a-chip devices. Though regenerative
medicine technologies may be developed or
used internationally, the policy options we
identified represent possible actions U.S.
policymakers and stakeholders could take.
Literature search
In the course of our review, we conducted a
literature search of key technologies for
curing human disease and restoring bodily
functions using search terms including
“regenerative medicine,” “bioprinting,” and
“organs,” among other keywords relevant to
technologies for regenerative medicine. We
also conducted a broad search of materials
published within the last 10 years, including
scholarly articles and government reports.
From these searches, we identified and
selected relevant articles to include in our
review. We used the results of our literature
review to inform our findings as well as
identify experts to interview or invite to
participate in our expert meeting.

Regenerative Medicine GAO-23-105430 43
Interviews
We interviewed federal agency officials and
researchers as well as nonfederal experts with
a diverse set of perspectives on the science
and application of these technologies. The
federal experts included individuals from FDA,
the National Institutes of Health, Department
of Defense, and National Institute of
Standards and Technology (NIST). We also
interviewed experts from technology
companies, universities, and research
institutes that use or develop regenerative
medicine technologies and representatives
from national advocacy organizations, such as
the American Society of Gene and Cell
Therapy and the Alliance for Regenerative
Medicine.
Expert meeting
To address all of our objectives, we also held
a 3-day expert meeting on April 13, 19, and
22, 2022. This meeting was held with
assistance from the National Academies and
was divided into six sessions: (1) emerging
regenerative medicine technologies; (2)
regulatory challenges for new regenerative
medicine technologies; (3) manufacturing and
standardization challenges in regenerative
medicine; (4) social, economic, and ethical
implications of emerging regenerative
medicine technologies; (5) translational
hurdles for emerging regenerative medicine
technologies; and (6) potential policy options
that could help address technology limitations
and other challenges.
56
56
This meeting of experts was planned and convened with
assistance from the National Academies to better ensure that a
breadth of expertise was brought to bear in its preparation.
We selected meeting participants based on
their expertise in at least one area related to
our objectives. We provided the National
Academies staff with descriptions of the
expertise needed by expert meeting
participants. From this information, the staff
provided an initial list of potential participants
for the expert meeting. We reviewed the list
and provided an additional list of experts
based on our review of the literature.
In addition to evaluating experts on the basis
of their expertise, we evaluated them for any
conflicts of interest. A conflict of interest was
considered to be any current financial or
other interest, such as an organizational
position, that might conflict with the service
of an individual because it could (1) impair
objectivity or (2) create an unfair competitive
advantage for any person or organization. Of
the 18 experts who participated in the expert
meeting, some were affiliated with
companies, government agencies,
universities, or public-private partnerships.
We took these affiliations into consideration
as potential conflicts of interest when
conducting our analysis and preparing our
report. We determined that these experts’
affiliations were unlikely to bias our overall
reporting.
Policy options
Based on our research, we developed a series
of policy options. These are not listed in any
particular order, nor are they inclusive of all
possible policy options. Policy options are
intended to represent possible options
policymakers can take to address a policy
However, all final decisions regarding meeting substance and
expert participation were the responsibility of GAO.
Regenerative Medicine GAO-23-105430 44
objective. We consider policymakers to
include Congress, federal agencies, state and
local governments, academia, and industry.
For each policy option, we discussed potential
opportunities and considerations. We limited
policy options to those that fit the objective
and fell within the report scope.
To develop our policy options, we compiled a
list of possible options over the course of our
work based on review of the literature,
interviews with experts, and our expert
meeting. We further refined and assessed
these options to ensure they were adequately
supported by the evidence we collected,
could be feasibly implemented, and fit into
the overall scope of our work. We then
analyzed the information we collected to
identify potential benefits and considerations
of implementing each policy option. The
policy options and analyses were supported
by documentary and testimonial evidence.
We conducted our work from September
2021 to July 2023 in accordance with all
sections of GAO’s Quality Assurance
Framework that are relevant to technology
assessments. The framework requires that we
plan and perform the engagement to obtain
sufficient and appropriate evidence to meet
our stated objectives and to discuss any
limitations to our work. Consistent with our
quality assurance framework, we provided
the relevant agencies and experts with a draft
of our report and solicited their feedback,
which we incorporated as appropriate. We
believe that the information and data
obtained, and the analysis conducted, provide
a reasonable basis for any findings and
conclusions in this product.
Regenerative Medicine GAO-23-105430 45
Appendix II: Expert Participation
We convened a 3-day meeting of 18 experts with assistance from the National Academies of
Sciences, Engineering, and Medicine to inform our work on regenerative medicine technologies;
the meeting was held virtually on April 13, 19, and 22, 2022. The experts who participated in this
meeting are listed below. Some of these experts gave us additional assistance throughout our
work, including eight experts who provided additional assistance during our study by sending
material for review or participating in interviews and the experts who reviewed our draft report
for accuracy and provided technical comments.
Guillermo Ameer
Northwestern University
Anthony Axtala
Wake Forest Institute for Regenerative
Medicine
Glenn Cohen
Harvard Law School
Kurt Gunter
Athenex
Kelvin Lee
University of Delaware; National Institute for
Innovation in Manufacturing
Biopharmaceuticals
Tim Miller
Forge Biologics
Richard McFarland
Advanced Regenerative Manufacturing
Institute; Standards Coordinating Body for
Gene, Cell, and Regenerative Medicines and
Cell-Based Drug Discovery
Maria Millan
California Institute for Regenerative Medicine
Mahendra Rao
PanCELLa
Liz Richardson
Pew Charitable Trusts
David Ridley
Duke University
Derek Robertson
The Maryland Sickle Cell Disease Association
Krishnendu Roy
Georgia Tech Cell Manufacturing
Technologies
Kris Saha
University of Wisconsin-Madison
Kevin Schulman
Stanford University
Sohel Talib
California Institute for Regenerative Medicine
Kathy Tsokas
Janssen Inc. Canada
James Yoo
Wake Forest Institute for Regenerative
Medicine
Regenerative Medicine GAO-23-105430 46
Appendix III: GAO Contact and Staff Acknowledgments
GAO contact
Karen L. Howard, PhD, Acting Chief Scientist and Director, Science, Technology Assessment, and
Analytics (STAA), at (202) 512-6888 or [email protected]
Staff acknowledgments
In addition to the contact named above, the following STAA staff made key contributions to
this report:
Sarah Harvey, MS, Assistant Director
Cindy Korir-Morrison, PhD, Analyst-in-Charge and Senior Biological Scientist
Kristin Hook, PhD, Biological Scientist
Eric D. Lee, PhD, Senior Biological Scientist
These staff also contributed to this work:
Nora Adkins, Senior Attorney
Virginia Chanley, PhD, Senior Design Methodologist
Jehan Chase, Senior Attorney
Ailene Edwards, Intern
Kaitlin Farquharson, Senior Attorney
John Karikari, PhD, Assistant Director, Economist
Flora Ngo, Intern
Emily Quick-Cole, Intern
Joe Rando, Visual Communications Analyst
Ben Shouse, MS, Communications Analyst
Walter Vance, PhD, Assistant Director

Regenerative Medicine GAO-23-105430 47
GAO’s Mission
The Government Accountability Office, the audit, evaluation, and investigative arm of Congress,
exists to support Congress in meeting its constitutional responsibilities and to help improve the
performance and accountability of the federal government for the American people. GAO
examines the use of public funds; evaluates federal programs and policies; and provides analyses,
recommendations, and other assistance to help Congress make informed oversight, policy, and
funding decisions. GAO’s commitment to good government is reflected in its core values of
accountability, integrity, and reliability.
Obtaining Copies of GAO Reports and Testimony
The fastest and easiest way to obtain copies of GAO documents at no cost is through GAO’s
website (https://www.gao.gov). Each weekday afternoon, GAO posts on its website newly
released reports, testimony, and correspondence. To have GAO e-mail you a list of newly posted
products, go to https://www.gao.gov and select “E-mail Updates.”
Order by Phone
The price of each GAO publication reflects GAO’s actual cost of production and distribution and
depends on the number of pages in the publication and whether the publication is printed in color
or black and white. Pricing and ordering information is posted on GAO’s website,
https://www.gao.gov/ordering.htm.
Place orders by calling (202) 512-6000, toll free (866) 801-7077, or TDD (202) 512-2537.
Orders may be paid for using American Express, Discover Card, MasterCard, Visa, check, or money
order. Call for additional information.
Connect with GAO
Connect with GAO on Facebook, Flickr, Twitter, and YouTube.Subscribe to our RSS Feeds or E-mail
Updates. Listen to our Podcasts and read The Watchblog.Visit GAO on the web at
https://www.gao.gov.
To Report Fraud, Waste, and Abuse in Federal Programs
Contact: Website: https://www.gao.gov/fraudnet/fraudnet.htm
Automated answering system: (800) 424-5454 or (202) 512-7470
Congressional Relations
A. Nicole Clowers, Managing Director, [email protected], (202) 512-4400,
U.S. Government Accountability Office, 441 G Street NW, Room 7125, Washington, DC 20548
Public Affairs
Chuck Young, Managing Director, Young[email protected], (202) 512-4800
U.S. Government Accountability Office, 441 G Street NW, Room 7149, Washington, DC 20548
Strategic Planning and External Liaison
Stephen Sanford, Managing Director, [email protected], (202) 512-9715
U.S. Government Accountability Office, 441 G Street NW, Room 7B37N, Washington, DC 20548
