
Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 1 of 12
MDCG 2022-10
Q&A on the interface between
Regulation (EU) 536/2014 on clinical trials
for medicinal products for human use (CTR)
and Regulation (EU) 2017/746 on in vitro
diagnostic medical devices (IVDR)
May 2022
This document has been endorsed by the Medical Device Coordination Group
(MDCG) and the Clinical Trial Expert Group (CTEG). The MDCG and the CTEG are
composed of representatives of all Member States and are chaired by a
representative of the European Commission.
The document is not a European Commission document and it cannot be regarded
as reflecting the official position of the European Commission. Any views expressed
in this document are not legally binding and only the Court of Justice of the European
Union can give binding interpretations of Union law.

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 2 of 12
Introduction
This Q&A intends to clarify certain interfaces between the Regulation (EU) No 536/2014 on
clinical trials for medicinal products for human use (CTR) and Regulation (EU) 2017/746 on
in vitro diagnostic medical devices (IVDR)
1
. It was developed by clinical trials experts from
Clinical Trials Facilitation and Coordination Group (CTFG) and in vitro diagnostics experts
from the IVD sub-group of the Medical Device Coordination Group (MDCG)
2
. It has been
adopted by CTFG, the Clinical Trials Expert Group of the European Commission and
endorsed by the MDCG.
The need for interoperability of databases for clinical trials with medicinal products and
medical devices is laid down in the new Regulations, however, no further details on the
interface of IVDR and CTR are in the legislation, e.g. the requirements for assays used in the
context of clinical trials.
Assays used in clinical trials may range from CE marked in vitro diagnostic medical devices
(IVDs) to trial- or medicinal product-specific assays that are not always meant to be
developed as IVDs. The need to clarify requirements for these assays lead to the Q&A and
the concept of the “medical purpose of an assay in a clinical trial” as a specific setting.
The CTR created a new framework for the assessments of initial clinical trial applications and
applications for substantial modifications. The assessment procedure under the CTR will be
driven by strict timelines and will require close cooperation between Member States
concerned in multi-country trials. The aim of this Q&A is to support these coordinated
assessments by providing clarification on the regulatory status of assays performed on
human samples used in the context of clinical trials as well as on the regulatory expectations
toward the clinical trial sponsors. The overall aim is to support the conduct of clinical trials
using diagnostic assays, including combined trials for the development of companion
diagnostics (CDx).
Non-interventional studies, defined as clinical studies other than a clinical trial in Article 2 (4)
of the CTR are out of scope of both the CTR (as per Article 1) and this document. However,
IVDs used in non-interventional clinical studies are subject to the IVDR.
The IVDR lays down rules concerning the “placing on the market”, “making available on the
market” or “putting into service” of IVDs and accessories for an IVD. It further outlines the
requirements for performance studies concerning IVDs and accessories for an IVD
conducted in the Union.
1
Regulation (EU) 2017/746 (OJ L 117, 5.5.2017, p. 176) replaces Directive 98/79/EC (OJ L 331, 7.12.1998, p. 1)
on in vitro diagnostic medical devices as of 26 May 2022. Regulation (EU) 2017/746 maintains many of the
legislative principles of Directive 98/79/EC e.g. regarding general definitions and CE-marking after conformity
assessment before placing medical devices on the EU market. Regulation (EU) 2017/746 introduces several
important changes including new definitions, Unique Device Identification (UDI) system for medical devices, and
new principles of classification of IVDs. Please note that Regulation (EU) 2022/112 (OJ L 19, 28.1.2022, p. 3)
amends Regulation (EU) 2017/746 with respect to transitional provisions for certain in vitro diagnostic medical
devices and the deferred application of conditions for in-house devices.
2
Expert group responsible for overseeing the implementation of Regulation (EU) 2017/746, set up according to
Art. 103 of Regulation (EU) 2017/745 and Art. 98 of Regulation (EU) 2017/746.

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 3 of 12
When reference is made to IVDs in the following text, this also includes CDx, as the
requirements do not differ from a clinical trials perspective. Information is included on the use
of in-house IVDs in the context of clinical trials.
The questions and answers below specifically addresses the use of assays in the framework
of clinical trials conducted under the CTR, in line with requirements of the IVDR and should
not be understood to apply beyond it.
The same issue might be covered in multiple questions, the reader is therefore encouraged
to read the entire document.
For guidance documents issued by the MDCG regarding IVDs refer to the European
Commission website.
For guidance on clinical trials please refer to EudraLex Vol. 10.
Glossary
CE marking: According to Article 2 (35) IVDR, ‘CE marking of conformity’ or ‘CE marking’
means a marking by which a manufacturer indicates that a device is in conformity with the
applicable requirements set out in this Regulation and other applicable Union harmonisation
legislation providing for its affixing
Performance Study Plan: According to Article 2 (43) IVDR ‘performance study plan’ means
a document that describes the rationale, objectives, design methodology, monitoring,
statistical considerations, organisation and conduct of a performance study;
Clinical Trial Application (CTA): According to CTR Article 5 or Article 11 an application for
authorisation submitted to the EU Clinical Trials Portal and Database
Clinical Trial Regulation (CTR): Regulation (EU) No 536/2014 (CTR) of 16 April 2014 on
clinical trials on medicinal products for human use, and repealing Directive 2001/20/EC, OJ L
158, 27.5.2014, p. 1. The expected application date of the CTR is 30 January 2022.
Companion diagnostic (CDx): Article 2 (7) IVDR’ means a device which is essential for the
safe and effective use of a corresponding medicinal product to:
(a) identify, before and/or during treatment, patients who are most likely to benefit
from the corresponding medicinal product; or
(b) identify, before and/or during treatment, patients likely to be at increased risk of
serious adverse reactions as a result of treatment with the corresponding medicinal
product;
Combined Trial: For the purpose of this document a combined trial is a simultaneous
investigation of a medicinal product (clinical trial authorised under the CTR) and an IVD
(clinical performance study). The combined trial is subject to requirements of both the Clinical
trial legislation (CTR when this applies) and the IVD legislation (IVDR when this applies).

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 4 of 12
Device for performance study according to IVDR Article 2 (45) means a device intended by
the manufacturer to be used in a performance study. A device intended to be used for
research purposes, without any medical objective, shall not be deemed to be a device for
performance study
Stratification: Within clinical trials, a method used in the randomization to ensure equal
distribution of chosen variables between treatment arms
In-house IVD: An IVD manufactured and used within the same health institution as outlined
in IVDR Article 5 (5). Health institution is defined in IVDR Article 2 (29).
Instructions for use according to Article 2 (14) IVDR means the information provided by the
manufacturer to inform the user of the device's intended purpose and proper use and of any
precautions to be taken
Intended purpose according to Article 2 (12) IVDR means the use for which a device is
intended according to the data supplied by the manufacturer on the label, in the instructions
for use or in promotional or sales materials or statements or as specified by the manufacturer
in the performance evaluation
Interventional clinical performance study according to Article 2 (46) IVDR means a clinical
performance study where the test results may influence patient management decisions
and/or may be used to guide treatment
Clinical Trial according to Article 2 (2) CTR, means a clinical study which fulfils any of the
following conditions: (a) the assignment of the subject to a particular therapeutic strategy is
decided in advance and does not fall within normal clinical practice of the Member State
concerned; (b) the decision to prescribe the investigational medicinal products is taken
together with the decision to include the subject in the clinical study; or (c) diagnostic or
monitoring procedures in addition to normal clinical practice are applied to the subjects. By
definition, all clinical trials are “interventional” For clarification on the differentiation between
clinical trial and non-interventional study, reference is also made to Question 1.5 in the Q&A
published on EudraLex vol. 10.
In vitro diagnostic medical device (IVD) is a medical device as defined in point (1) of
Article 2 of Regulation (EU) 2017/745 on medical devices and, more specifically, according
to Article 2 (2) IVDR, any medical device which is a reagent, reagent product, calibrator,
control material, kit, instrument, apparatus, piece of equipment, software or system, whether
used alone or in combination, intended by the manufacturer to be used in vitro for the
examination of specimens, including blood and tissue donations, derived from the human
body, solely or principally for the purpose of providing information on one or more of the
following: (a) concerning a physiological or pathological process or state; (b) concerning
congenital physical or mental impairments; (c) concerning the predisposition to a medical
condition or a disease; (d) to determine the safety and compatibility with potential recipients;
(e) to predict treatment response or reactions; (f) to define or monitoring therapeutic
measures. Specimen receptacles shall also be deemed to be in vitro diagnostic medical
devices;

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 5 of 12
IVD manufacturer according to Article 2 (23) IVDR means a natural or legal person who
manufactures or fully refurbishes a device or has a device designed, manufactured or fully
refurbished, and markets that device under its name or trade mark
IVDR: Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April
2017 on in vitro diagnostic medical devices and repealing Directive 98/79/EC and
Commission Decision 2010/227/EU, OJ L 117, 5.5.2017, p. 176.
Making available on the market according to Article 2 (20) IVDR, means any supply of a
device, other than a device for performance study, for distribution, consumption or use on the
Union market in the course of a commercial activity, whether in return for payment or free of
charge;
Performance study according to Article 2 (42) IVDR, means a study undertaken to establish
or confirm the analytical or clinical performance of a device
Placing on market according to Article 2 (21) IVDR, means the first making available of a
device, other than a device for performance study, on the Union market
Putting into service according to Article 2(22) IVDR, means the stage at which a device,
other than a device for performance study, has been made available to the final user as
being ready for use on the Union market for the first time for its intended purpose;
Substantial modification according to Article 2.2.13 CTR means any change to any aspect
of the clinical trial which is made to ongoing clinical trials and which is likely to have a
substantial impact on the safety or rights of the subjects or on the reliability and robustness
of the data generated in the clinical trial.
Questions and Answers
1. Question: Regarding IVDs that are currently on the market on the basis of
Directive 98/79/EC, do they need to undergo conformity assessment under the
IVDR to remain on the market under that Regulation?
Answer: The IVDR does not foresee a recognition of Directive marketed IVDs
but lays down various transitional provisions for IVDs compliant with Directive
98/79/EC. For more details on transitional provisions see the EU Commission
website.
2. Question: Do all IVDs used in a clinical trial require CE marking at the time of
the clinical trial?
Answer: Not all. An IVD used in a clinical trial needs to be compliant with the
in vitro diagnostic medical device legislation. This implies that the IVD either
has the CE marking for the intended purpose, or is an in-house IVD (see Q3)
or is a device for a performance study according to the IVDR ongoing in
parallel (see Q6, Q7). In-house IVDs and devices for performance study are
not required to bear CE-marking.

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 6 of 12
3. Question: Can I use an in-house IVD in a clinical trial?
Answer: Yes. This remains possible.
With the exception of the relevant general safety and performance
requirements set out in Annex I, the requirements of the IVDR shall not apply
to devices manufactured and used only within health institutions established in
the Union, provided that all of the conditions of Article 5(5) are met.
In-house IVDs might also be regulated in national legislation and it is
necessary to check with the national competent authorities responsible for
performance studies, which requirements apply.
4. Question: Are all assays used in a clinical trial subject to the IVD legislation?
Answer: No, not all assays, but those that fulfil the definition of an IVD are
subject to the IVD legislation. Such assays may have a medical purpose
within the clinical trial, e.g. when they guide medical management decisions or
follow-up (see Q6).
The IVD Directive 98/79/EC and the IVDR lay down rules concerning the
placing on the market, making available on the market or putting into service
of IVD for human use and accessories for such devices in the European
Union. In addition, the IVDR lays down rules specific to devices for
performance studies and in-house devices.
5. Question: When does an assay used on human samples qualify as an IVD in a
clinical trial?
Answer: An assay is considered an IVD if the manufacturer assigns an
intended purpose that fulfils the definition of an IVD according to IVDR Article
2 (see Q6). Where a clinical trial sponsor assigns a medical purpose to an
assay in the context of the clinical trial in a way that the assay fulfils the
definition of an IVD according to IVD Regulation 2017/746 Article 2, the
clinical trial sponsor may assume the role of a manufacturer under the IVDR
3
.
In this role, it is up to the clinical trial sponsor to determine the regulatory
status of the assay based on the planned use in the clinical trial. In case of
doubt, consultation of the respective NCAs for IVDs and for clinical trials (with
medicinal products) is recommended.
3
See Article 16(1) IVDR.
Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 7 of 12
6. Question: What are examples of assays used in clinical trials which are IVDs?
Answer: Some examples include assays providing information for clinical trial
related medical management decisions (typically to select patients for
enrolment in the trial, assign patients to a treatment arm, etc.) and/or may be
used to guide follow up measures during and beyond the clinical trial. This
would, for example, not be the case, in settings where all trial participants are
tested irrespective of treatment arm or medical management and the analysis
of impact is conducted retrospectively and where medical management is not
impacted by assay results.
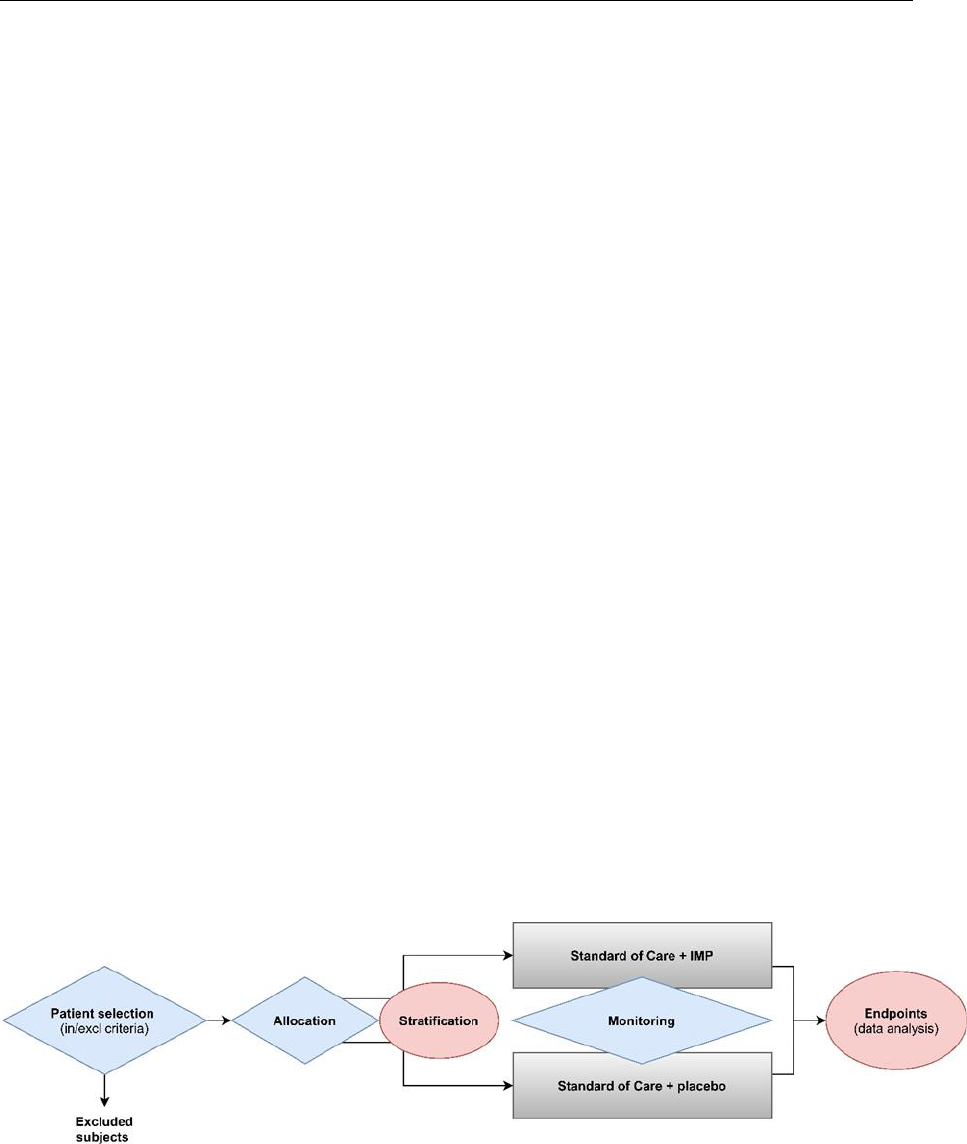
Figure 1 visualises, as an example, the flow of a blinded clinical trial with two
treatment arms, where the key processes for which assays might be utilised
are highlighted. The processes in blue are considered to be used for medical
management decisions of trial subjects. These include assays used for
inclusion and exclusion of subjects, treatment allocation as well as monitoring
the safety and efficacy of the treatment during the trial.
The processes in pink are likely not to impact the medical management of the
trial subjects. These include stratification and endpoint analysis or other
exploratory assays for which correlation with clinical parameters is
investigated retrospectively without impact on patient treatment (medical
purpose). In relation to endpoints, it is important to acknowledge that these
assays may be considered IVDs in future clinical trials (e.g. used for allocation
or monitoring). Where this development is predictable, the assay should be
developed and validated in compliance with the applicable requirements of
Annex I of the IVDR as an IVD from the beginning. Importantly, in most cases,
the assay will also be utilised during the trial as part of the monitoring of the
trial subjects, which implies need for compliance with IVDR requirements.
Fig. 1 Simplified examples of use of assays on human samples in a clinical trial.
Assays marked in blue (diamonds) are considered to be assays which will likely be
considered IVDs as they are used for medical management decisions of trial subjects
within the trial. The processes in yellow pink (ellipses) are considered to likely not to
impact the medical management of the trial subjects and therefore would not have a
medical purpose in the trial.

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 8 of 12
7. Question: In which situations may a non-CE marked IVD (or an IVD that is CE
marked for another intended purpose) be used in a clinical trial for a medical
purpose according to the IVDR?
Answer: Note that using a CE marked IVD outside its prescribed intended
purpose is not covered by the CE marking provided to that device and renders
it a non-CE marked IVD in the context of this clinical trial.
Where a non-CE marked IVD is used for a medical purpose in a clinical trial
the IVD must either be
a device for performance study (it is not acceptable to use an
investigational IVD without evaluating its performance), or
an in-house IVD (see also Q3).
If a non-CE marked assay is used for a medical purpose in a way that the
assay fulfils the definition of an IVD according to Article 2 of the IVDR, outside
of the abovementioned settings, this is not compliant with the IVDR. It is the
responsibility of the clinical trial sponsor to ensure compliance with relevant
provisions of Union and national law, including the IVDR.
If performance data is generated for an IVD outside of a performance study it
might be insufficient for a future performance evaluation, and thus further
studies might be required to generate sufficient clinical evidence.
8. Question: Is a use in research sufficient justification for an assay to be defined
as Research Use Only?
Answer: No. Where an assay, instrument, apparatus, appliance material or
other article, including software is intended for research use only and is
assigned a medical purpose by the clinical trial sponsor in the clinical trial
protocol in a way that the assay fulfils the definition of an IVD according to
IVDR Article 2, it becomes an IVD and can no longer be considered a
research use only assay. It becomes regulated under the IVDR (see Q4-Q6).
9. Question: Can an EU core lab facility
4
that is part of a health institution
established in the Union and has developed an in-house IVD receive samples
from several clinical trial sites for analysis, or does the in-house IVD device
have to be co-located with the clinical trial site?
Answer: In-house devices can be used as long as the provisions of the IVDR
and relevant national legislations are satisfied, which include notably, that the
device must be manufactured and used within a health institution established
in the EU and the device is not transferred to another legal entity. In-house
4
A central laboratory to analyse samples from multiple sites within a clinical trial

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 9 of 12
IVD must fulfil relevant general safety and performance requirements set out
in Annex I.
5
Samples can travel and be analysed by means of in-house devices based in a
different location than the trial site facilitating a core lab facility approach. The
in-house concept applies to the device itself, which must be manufactured and
used within the responsible health institution.
10. Question: Is there a common EU procedure for combined trials?
Answer: No, there is no dedicated procedure foreseen in the Regulations and
therefore, currently, no harmonized procedure is in place. However, where a
single combined trial will serve as the clinical performance study for the device
and the clinical trial of the medicinal product, the study documentation
required for submissions (if applicable) may be partially overlapping. Not all
clinical performance studies have to be submitted to the regulatory authorities
(see Q14). Sponsors of these clinical trials are encouraged to consult national
guidance documents and contact National Competent Authorities prior to
clinical trial submission.
11. Question: Which IVDs should be listed in the cover letter of the clinical trial
application?
Answer: Only those IVDs should be mentioned in the cover letter that are
specifically required by the protocol to be used to achieve the objectives of the
clinical trial. The information provided should permit identification of the IVD
being used and whether it is CE-marked for the planned use. Evidence of CE-
marking generally does not have to be provided in the CTA, but it is the
responsibility of the clinical trial sponsor to determine whether the device is
used in the clinical trial in line with the manufacturer’s intended purpose.
12. Question: Which information needs to be provided in the clinical trial
application
6
, if an assay with medical purpose that qualifies as an IVD is used in
a clinical trial where the assay does not have a CE marking for the intended
purpose (including in-house assays)?
Answer: According to Annex I.B.7 (i) of the CTR, the clinical trial sponsor
shall include in the Cover letter the list of medical devices which are to be
investigated in the clinical trial but which are not part of the investigational
medicinal product or products, together with a statement as to whether the
5
The health institution must provide reasoned justification on which requirements are not fully met according to
article 5(f) (iii) IVDR.
6
In this document ‘clinical trial application’ covers both the initial trial application and applications for substantial
modifications to add an assay with a medical purpose to an ongoing trial

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 10 of 12
medical devices are CE-marked for the intended purpose. This should be
understood to refer to medical devices (including IVDs) which are specifically
required by the protocol to be used to achieve the objectives of the trial.
Use of an IVD in a clinical trial may meet the definition of a device for
performance study. (See glossary ‘device for performance study’ and
‘performance study’)
For each device used in the clinical trial that meets the definition of a device
for performance study, the sponsor of the clinical trial should comment on the
need for notification or application for the clinical performance study
conducted in parallel with the clinical trial, based on the applicable IVD
legislation. Reference should be made to EUDAMED Single Identification
Number (SIN) and/or National Competent Authority reference numbers (if
available) for planned or submitted performance studies. To facilitate bridging
between clinical trial and performance study, the IVD being used should be
identified in the cover letter and protocol of the clinical trial.
Where the IVD manufacturer is supporting the clinical trial, which is also a
performance study, the clinical trial sponsor should obtain a statement from
the IVD manufacturer that the device for performance study in question
conforms to the general safety and performance requirements laid down in
Annex I of the IVDR, apart from the aspects covered by the clinical
performance study and that, with regards to those aspects, every precaution
has been taken to protect the health and safety of the subject.
Where the clinical trial sponsor is also the manufacturer of the IVD or
assumes the role as manufacturer of the IVD according to Article 16 IVDR, the
clinical trial sponsor must draw up their own statement as above. In case the
study falls under IVDR Art 58 (1) or (2), it must be designed, authorised,
conducted, recorded and reported in accordance with IVDR Art. 58-77 and
Annex XIV.
In the CTA cover letter, the availability of this statement should be confirmed,
and the aspects that are covered in performance study as part of the clinical
trial should be listed. The statement is to be kept with the sponsor’s trial
master file (TMF) and investigators’ site file (ISF) and made available for
inspections or at the request of NCAs.
In case of confidential information in the description of the performance
studies, a redacted cover letter can be submitted, as necessary.
In addition, further details on compliance with IVDR and Annex I of the IVDR
can be provided in the protocol, according to CTR Annex I 17. a)-f).

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 11 of 12
13. Question: Which information needs to be provided in the clinical trial
application for an in-house IVD?
Answer: In principle, in the case of in-house IVDs, the sponsor should provide
the name of the health institution where the IVD is manufactured and used
and a link to their IVDR Art 5(f) declaration. It is recommended to document
this aspect in the harmonised template document
7
.
The IVD analysis must be carried out in a laboratory complying with
ISO15189, or, where applicable, with national provisions, in line with IVDR
Article 5(5)(c).
In addition, further details on compliance with IVDR Article 5(5) and Annex I of
the IVDR can be provided in the protocol, according to CTR Annex I 17. a)-f).
Please note that Regulation (EU) 2022/112 amends the IVDR to defer the
application of certain conditions for in-house devices.
14. Question: What are the notification/application requirements for the
performance study according to the IVDR?
Answer: Where the performance study is either an interventional clinical
performance study or involves risks for the subjects of the study an application
is necessary. Additional reporting and documentation obligations apply
according to Art. 58 (1) and (2) IVDR.
Where the IVD is being used according to its intended purpose outlined in the
instructions for use, Article 70 may apply. The submission requirements from
the IVDR perspective will depend on the particulars of the device in question
and the design of the performance study (Article 57, 58 and 70 IVDR).
15. Question: What are the respective responsibilities of the clinical trial sponsor
and IVD manufacturer including in the case of co-development and use in a
combined trial?
Answer: Clinical trial sponsor requirements are defined in Article 2.2.14 CTR:
The sponsor is responsible for the initiation and management of the clinical
trial, including the selection and use of IVDs. The sponsor is responsible for
the overall compliance of products used in the clinical trial with the CTR and
other relevant EU and National legislation. Clinical trial sponsors are reminded
of their obligations according to ICH-GCP E6 (R2) to document the
competence of a laboratory to perform a certain test to support the reliability of
results. This information should be kept at the sponsor’s Trial Master File
(TMF) and, when applicable, at the investigator’s site file. Site suitability in
7
Available on EudraLex vol. 10.

Medical Device Coordination Group Document MDCG 2022-10
Clinical Trials Expert Group Document
Page 12 of 12
accordance with Art 50 and Annex I N67 of the CTR should be documented.
As good practice recommendation, this should take place by using the
harmonised template document.
In case of a clinical trial where the IVD is used outside its intended purpose
the clinical trial sponsor assumes the obligations incumbent on manufacturers
according to Article 16(1) of the IVDR
8
.
It is the intended purpose, which determines the applicability of the IVDR for
an assay and not the intention (or not) of obtaining a CE-mark at a later stage.
Where the assay qualifies as an IVD under the IVDR a natural or legal
person
9
needs to take responsibility for the safety and performance of the IVD
device.
16. Question: In situations where it is procedurally feasible and where the same
ethics committee is responsible for evaluating the clinical trial AND
performance study aspects of a combined trial – Would there be a legal
requirement for two separate ethics opinions, or could the outcome be a single
document for both aspects?
Answer: According to IVDR Art. 2 (59) and Art 58 (3), and CTR Art. 2.2.11,
recital (18) and Art. 4, the ethics committees operate according to national
law. It is up to the Member States to organize the work of the ethics
committees and it is up to the Member States to ensure that the procedures
for the review by the ethics committees are compatible with the clinical trial or
conformity assessment procedures in the CTR and IVDR respectively.
However, in situations where it is procedurally feasible and where the same
ethics committee is responsible for evaluating the clinical trial and
performance evaluation aspects of a combined trial, there is no legal
requirement for two separate ethics opinions. The outcome could be a single
document for both aspects, provided this is permissible under national law of
the member state.
At the same time, in accordance with Art 9 of the CTR, Member States shall
ensure that the persons validating and assessing the application do not have
conflicts of interest, are independent of the sponsor, of the clinical trial site and
the investigators involved and of persons financing the clinical trial, as well as
free of any other undue influence. Member States shall ensure that persons
admitting and assessing the application have no financial or personal
interests, which could affect their impartiality.
8
See also Q5.
9
Which might be the manufacturer, sponsor of the clinical trial, the developer of the investigational medicinal
product, a health institution, a research group etc.
