
ASHP Guidelines for the Management of
Investigational Drug Products
Purpose
The purpose of these guidelines is to describe a standard-
ized approach for the management of investigational drug
products by the clinical research pharmacy, pharmaceuti-
cal industry, and cooperative and research network groups.
The scope of these guidelines includes the receipt, account-
ability, storage, handling, preparation, dispensing, and fi-
nal disposition of investigational drug products to ensure
inspection readiness and compliance with regulations as
provided in the Code of Federal Regulations (CFR), 21
CFR, Part 312,
1
as well as International Conference on
Harmonisation of Technical Requirements for Registration
of Pharmaceuticals for Human Use (ICH) E6 Good Clinical
Practice
2
(GCP) (described in 21 CFR Part 312, section 120)
and Good Manufacturing Practice
3
(GMP) (described in 21
CFR Part 211), and the approved clinical study protocols.
These guidelines will facilitate the adoption of best practices
by new and established clinical research pharmacies (e.g.,
investigational drug service [IDS]) in collaboration with the
pharmaceutical industry for the optimal management of in-
vestigational drug products. The ultimate goals of standard-
izing the management of investigational drug products are
to improve patient safety, improve efficiency, and provide
robust clinical data that allow new and innovative medica-
tions to reach the patients who need them.
Value of a Clinical Research Pharmacy
Since 1962, the Kefauver-Harris Amendments
4
to the
Federal Food, Drug and Cosmetic Act have required drug
manufacturers to prove that new drugs are safe and effective.
This requirement is the foundation for clinical studies as
they are known today. The pharmaceutical industry spends
billions of dollars each year to test investigational drug prod-
ucts for safety and efficacy, and it may take a decade or more
for a drug product to obtain market approval.
5
Thousands
of subjects have been asked to consent and take part in the
clinical research study process.
The current practice of the biopharmaceutical indus-
try is to manufacture, distribute, and monitor investigational
drug products for clinical research use while utilizing the
expertise of healthcare providers, especially pharmacists,
who manage the products at clinical study sites. It is impera-
tive that pharmacists participating in these studies have the
expertise to manage investigational drug products appropri-
ately. The clinical research pharmacist is a critical member
of the clinical study site team and has the expertise to under-
stand the special handling requirements of investigational
drug products.
6
Clinical research pharmacists understand the
importance of managing and properly documenting inves-
tigational product receipt, storage, dispensing, returns, and
final disposition. Failure to accurately document investiga-
tional product accountability can undermine the validity of
clinical study data, which could spur sponsors to halt a site’s
participation in current or future studies and result in a loss
of local patients’ access to clinical studies.
Clinical research pharmacy is a specialized area of
pharmacy practice that has evolved to meet the needs of the
clinical study sites, help ensure research participants’ safety,
and protect the integrity of clinical study data. Clinical re-
search pharmacists possess an expert working knowledge of
the clinical research study process, human subject protec-
tion, and national and local regulations governing drug re-
search. They are responsible for providing information to the
appropriate healthcare team members, including pharmacy
staff, who may be unfamiliar with the investigational drug
product, enabling them to correctly dispense it as described
in the clinical protocol and ensure its safe use.
Clinical Research Pharmacy Models
A clinical research pharmacy may be as simple as a part-time
pharmacist or as complex as a team of dedicated clinical re-
search pharmacists, technicians, and coordinators. It most
commonly is part of the larger pharmacy organization (e.g.,
the hospital pharmacy). In rare cases, it may be a freestand-
ing entity. As the complexity and volume of clinical studies
at an institution increases, the decision to create a dedicated
clinical research pharmacy with highly trained and special-
ized personnel and management can be made.
When establishing a clinical research pharmacy, the
appropriate institutional metrics (e.g., staffing-to-order vol-
ume ratios)
6,7
should be used to determine the appropriate
staffing, facility, and storage requirements needed to manage
investigational drug products. A comprehensive approach
will allow institutional leadership to fully support the cre-
ation of a clinical research pharmacy and should include a
review of the types and therapeutic areas of studies that will
be handled.
Each institution must also establish a funding model
to support the clinical research pharmacy. This model may
include (1) direct cost recovery from the sponsor–investiga-
tor, (2) indirect funding (e.g., based on institutional over-
head such as grants to conduct research), (3) foundation
underwriting of the research, or (4) institutions absorbing
the cost of the clinical research. Clinical research pharmacy
fees should be included at an early stage during the nego-
tiation of each clinical study budget. Investigational drug
products provided by sponsors at no cost to the clinical
study
site for use in clinical studies must not be charged to
study participants.
Within the organization, pharmacy leadership, the clin-
ical research pharmacist, institutional research leadership,
and investigators should establish the locations where re-
search participants will receive their care with the appropri-
ate pharmacy support. A review of pharmacy activities and
workflow for investigational drug product handling should
be performed for the following locations, as appropriate: (1)
inpatient units, (2) clinical research unit, (3) ambulatory care
or outpatient clinic setting, and (4) a combination of inpa-
tient and outpatient areas. The organization should develop
a mechanism to handle participants being treated at multiple
locations and facilities.
Research–Guidelines 645

646 Research–Guidelines
A dedicated team should be identified as the clinical
research pharmacy staff. In institutions with a large volume
of clinical studies, the clinical research pharmacy staff may
form an IDS with the appropriate management structure
depending on the practice environment. Members of the
clinical research pharmacy staff must have knowledge and
documented training on ICH GCP,
2
the Health Insurance
Portability and Accountability Act,
8
institutional review
board (IRB) review and protection of human subjects,
9
the
Belmont Report,
10
and all other competencies and policies
required by the institution or the pharmacy.
Facilities, Security, and Limited
Staff Access
According to GCP guidelines, the investigational drug prod-
uct should be stored in a secure location as specified by the
sponsor and in accordance with all applicable regulatory
requirements.
2
Some institutions may have separate rooms
for the storage of investigational drug products; others may
have a separate storage area within the pharmacy depart-
ment. In either case, the area should be secured (e.g., by key
or electronic lock), with entry restricted to clinical research
pharmacy or delegated staff.
Each institution must evaluate its ability to provide
secure, limited access to investigational drug products. A
clinical research pharmacy that does not have continuous
(24-hour/365-day) support to provide access to investiga-
tional drug products may mitigate the need for access by
utilizing regular pharmacy staff and considering on-call sup-
port by the clinical research pharmacists to supplement this
staff.
Temperature Control and Monitoring
Control and monitoring of investigational drug product stor-
age conditions are important for maintaining the integrity of
the products and for the safety of the research participants.
Documentation that proper storage conditions in the phar-
macy have been maintained must be available. The sponsor
shall determine and communicate acceptable storage tem-
peratures, storage conditions (e.g., protection from light),
and storage times for the products.
11
The clinical research
pharmacist must be able to document the temperature stor-
age conditions of the investigational drug products at all
times while managed by the pharmacy. Each pharmacy loca-
tion where investigational drug products are stored must be
monitored to ensure that proper temperature storage condi-
tions have been maintained.
Clinical research pharmacy and the pharmaceutical in-
dustry shall follow United States Pharmacopeia standards
for controlled temperature storage.
12
Before the start of ac-
tivities at the clinical study site, the sponsor should estab-
lish and communicate known allowable out-of-range tem-
peratures and the maximum allowable deviation time (i.e.,
acceptable time out of range) for the investigational drug
product. This information will enable the clinical research
pharmacy to make independent decisions without quarantin-
ing the investigational drug product supply for acceptable
excursions. An investigational drug product that does not
meet the allowable excursion parameters must be quaran-
tined under the proper storage conditions by the clinical
research pharmacy, and the sponsor and principal investiga-
tor (PI) must be notified. Prompt direction from the sponsor
regarding how to handle the quarantined product must be
adequately documented and kept in the site study file.
If the investigational drug product is sensitive to hu-
midity, the sponsor must work directly with the clinical
research pharmacy regarding monitoring requirements for
relative humidity.
All locations, including refrigerator, freezer, and
room-temperature areas, storing the investigational drug
product must have a temperature monitoring device or sys-
tem. In addition, the temperature monitoring system should
be calibrated at least annually to meet National Institute of
Standards and Technology standards.
13
Documentation of
the calibration of each device must be maintained and avail-
able for inspection. If an electronic temperature monitoring
system is not available, a calibrated manual thermometer
with the ability to capture daily maximum and minimum
temperatures should be used. A daily record of the maximum
and minimum temperatures should be maintained.
Equipment used to store investigational drug product
should be connected to a backup power supply in the event
of power failure. All refrigerators and freezers shall have
preventive maintenance completed annually. All calibration
and maintenance records should be archived per institutional
policy.
Site Qualification
The site qualification by the sponsor determines whether the
site meets the potential protocol requirements.
11
A pharmacy
assessment should be part of the sponsor’s site qualification.
Ideally, the sponsor’s agent should review the site’s policies
and procedures and applicable sponsor site qualification re-
quirements with the clinical research pharmacy staff. If the
sponsor provides a written report of the site qualification to
the site PI, a copy should also be forwarded to the clinical
research pharmacy. This report serves as the minutes of the
visit and agreement of the scope and services the clinical
research pharmacy may provide. If a report is not provided,
informal notes or minutes may be taken by clinical research
pharmacy personnel and kept on file.
Clinical Research Pharmacy Staff
Responsibilities
The clinical research pharmacy should be included in the
pre-IRB review for new clinical protocols. During this pre-
IRB review, the clinical research pharmacy should determine
the safety and feasibility of conducting the clinical research
study at the institution with respect to its policies, pharmacy
services, and the fees that will be charged for the services
provided. For this review, the clinical research pharmacist
should compile the following documents and information
from the clinical study site team and sponsor:
•
Clinical trial protocol, including the following:
•
Research participant location (inpatient, outpa-
tient, or both)
•
Number of research participants expected to be
enrolled at the site and number of dispensing
visits

Research–Guidelines 647
•
Duration of the study, including recruitment and
treatment periods
•
Randomization method (i.e., how participants
are randomized in the clinical trial)
•
Blinding (e.g., open label, single blind, double
blind, third-party blinding by clinical research
pharmacy)
•
Investigator’s brochure (known investigational drug
product information including safety data, any rele-
vant animal and human data, and the doses previously
studied; provided by sponsor)
•
Investigational drug product handling manual (phar-
macy manual), if available, which should include the
following:
•
Interactive response technology (IRT) (e.g., in-
teractive voice or Web response systems [IVRS/
IWRS]) instructions, if applicable
•
Description of investigational drug product (e.g.,
dosage forms, strength, packaging)
•
Investigational drug product and ancillary sup-
ply sourcing (e.g., provided by sponsor, sourced
locally by clinical research pharmacy)
•
Ancillary supplies required (e.g., filters, bags,
tubing, bag covers)
•
Supply of concomitant medications required
(e.g., adjunctive therapies, premedications)
•
A discussion should be raised during the
study setup to determine which concomi-
tant medications will be reimbursed and
which will be considered standard of care
•
Investigational drug product storage conditions
(e.g., temperature, humidity)
•
Special handling precautions (e.g., protect from
light, hazardous handling precautions)
•
Investigational drug product preparation or
dispensing information (e.g., diluents, stability
of reconstituted vial and solution for admin-
istration, dispensing in original container or
repackaging)
•
Handling of containers used in preparing dose
•
Handling of participant investigational drug
product returns
•
Administration of investigational drug product (e.g.,
flushing of infusion line, order of investigational drug
product administration in relation to other drugs ad-
ministered at the same visit, drug and food interac-
tions)
Pharmacist Listing on
Statement of Investigator
The decision whether to list a pharmacist on the Statement
of Investigator (Form FDA 1572) depends on the contribu-
tion the individual makes to the study.
14
If the individual will
make a direct and significant contribution to the data or is
directly involved in the treatment or evaluation of partici-
pants, he or she should be listed. If the individual will pro-
vide ancillary or intermittent contributions, he or she should
not be listed. A pharmacist who prepares investigational
drug product doses and maintains drug accountability for
multiple clinical studies that are conducted at an institution
would not be making a direct and significant contribution to
the data for a particular study; therefore, it would not be nec-
essary to list the pharmacist as a subinvestigator. However,
the pharmacist should be listed in the investigator’s study
records as an individual to whom specific responsibilities
have been delegated.
Delegation of Authority to Technicians
and Pharmacy Support Staff
Many sites utilize individuals who are not pharmacists to
perform some of the functions of the clinical research phar-
macy under the direction of a licensed pharmacist. Certified
pharmacy technicians or pharmacy coordinators may be del-
egated tasks, according to institutional policy, that do not re-
quire a pharmacist license. The clinical research pharmacist
is responsible for ensuring compliance with laws, rules, and
regulations regarding technician responsibilities. In many
states, a technician may only perform certain duties under
the direct supervision of a pharmacist, and the pharmacist is
ultimately responsible for the work performed by the techni-
cian; therefore, only the clinical research pharmacist should
sign the sponsor’s Delegation of Authority log.
Clinical Research
Pharmacy Staff Training
The clinical research pharmacist must be a participant in
the sponsor’s site initiation visit or the internal site initia-
tion meeting (for sponsor–investigator or cooperative group
studies). The clinical research pharmacist must participate in
the pharmacy-specific training session and have the oppor-
tunity to discuss the investigational drug product dispens-
ing logistics with the sponsor and clinical site study team.
The clinical research pharmacist should provide the spon-
sor with key policies and procedures and standard operating
procedures (SOPs) for review and approval, if not provided
at the site qualification visit. These may include but are not
limited to SOPs on training and delegation of tasks assigned
to pharmacy personnel regarding investigational drug prod-
uct handling, temperature monitoring, onsite investigational
drug product destruction, essential document retention, and
monitoring visit guidelines. Occasionally, sponsors conduct
the site initiation remotely via telephone or Internet confer-
ence. The clinical research pharmacist must be invited and
should participate in the pharmacy-specific training session.
At the site initiation visit, the pharmacy responsibili-
ties are determined and the Delegation of Authority form is
completed and signed by the clinical research pharmacist
and PI. Specific tasks need to be determined at study initia-
tion by the clinical research pharmacy and the clinical study
site team (e.g., which team member is accessing the IRT for
randomization, kit/bottle assignment). The clinical research
pharmacist should determine which qualified pharmacy staff
member will perform the delegated roles. The clinical re-
search pharmacist should also determine which pharmacy
staff will require electronic signatures (user name and pass-
words) for protocol systems such as IRT and coordinate with
the sponsor to obtain them.
All pharmacy staff who may dispense investigational
drug products should be trained in the proper dispens-
ing process and in GCP
2
as it relates to PI-delegated roles.
Training should be completed upon hire, and the information
should be reviewed periodically. Records of training must be

648 Research–Guidelines
maintained and made available for inspection by regulatory
agencies and sponsors.
Clinical research pharmacists and staff are required to
follow all relevant pharmacy practice standards, institution
policies and procedures, state and federal laws, and Joint
Commission or other accreditation standards, as well as GCP
guidance.
2
Clinical research pharmacy staff should maintain
all certifications and competencies specified by the pharmacy
department and the institution related to this requirement.
Formal sponsor training for every pharmacy staff
member for each protocol and all protocol amendments
is often not practical. Pharmacy staff should be trained to
refer to the most recent version of the study-specific dis-
pensing guidelines every time a delegated responsibility is
performed. This practice ensures that the most recent proce-
dures are used each time the investigational drug product is
prepared and dispensed. Therefore, study-specific training
by the sponsor is not indicated for all pharmacy staff. A phar-
macy signature log with staff initials should be maintained
and archived centrally by the pharmacy department for the
purpose of verifying study-related records.
Sponsors frequently request the curriculum vitae (CV)
of all pharmacy staff handling investigational drug product.
However, since the institution maintains licensure and cre-
dentialing information, it is appropriate for clinical research
pharmacy staff to only provide CVs to the sponsor if the
pharmacist is listed on Form FDA 1571 (Investigational
New Drug Application) or Form FDA 1572 (Statement of
Investigator).
1
The clinical research pharmacist should determine
whether interested parties can confirm pharmacist licensing
information at the individual state licensing office and, if so,
provide the sponsor with access information. It is not neces-
sary to provide proof of pharmacy technician certification.
Clinical Research Pharmacy Study Setup
The process of opening a new clinical research study should
include preparing dispensing guidelines, a model physician
order (prescription template), and a template of the dispens-
ing label. The process should also include creating a proto-
col-specific study drug entry and order set in the electronic
medical record as well as setting up a clinical research phar-
macy study file.
Dispensing guidelines should be prepared by the clini-
cal research pharmacist using the clinical protocol and any
other relevant information provided to the clinical research
pharmacy by the sponsor (e.g., investigational drug product
handling manual, site initiation visit materials, investiga-
tor’s brochure). It is the responsibility of the PI to ensure
that the research pharmacy has the most up-to-date versions
of all study documents. Dispensing guidelines describe the
protocol-specific functions that the clinical research phar-
macy staff must perform in order to adhere to the protocol
and complete all responsibilities delegated to the pharmacy
by the PI.
14
The dispensing guidelines are intended to opera-
tionalize the management of investigational drug products
for a specific protocol within the institution and are used as
a training tool by the pharmacy staff involved in the prepara-
tion and dispensing of the investigational drug product. A
sample of dispensing guidelines has been previously pub-
lished.
15
A draft version of the dispensing guidelines should
be prepared before the site initiation visit to allow the clinical
research pharmacist to identify the information that needs to
be obtained at the meeting and to ensure that the clinical site
will have all of the information needed before enrolling the
first participant into the study.
Dispensing guidelines should be revised as appropriate
after IRB approval of protocol amendments or other changes
to study procedures. The clinical research pharmacist should
maintain a system to document version control. Dispensing
guidelines should be available (in print or electronic for-
mats) to any pharmacy staff responsible for participant care.
The physician order template for the investigational
drug product should be prepared by the clinical research
pharmacist using the clinical protocol and other relevant
information provided by the sponsor.
5
In institutions that
use computerized provider order entry, the clinical research
pharmacist should ensure that an order set is available in the
system.
The investigational drug product should be entered
into the pharmacy computer dispensing systems, if avail-
able. A dispensing label template should be created to ensure
that information required by the clinical protocol is included
on the label and that the administration instructions are con-
sistent with the clinical protocol.
The dispensing label for the investigational drug prod-
uct must comply with all state and federal rules and regu-
lations as well as protocol requirements. The information
on the label should be consistent with the information and
instructions on the physician’s order as well as the original
drug container. The institution’s contact information should
be included on the label to aid in coordination of the par-
ticipant’s care by an outside institution. Special instructions
should be included on the dispensing label or provided as
auxiliary labels (e.g., food intake recommendations, storage
conditions).
If the clinical research pharmacy repackages the in-
vestigational drug product, the Federal Investigational
Drug Caution Statement —“Caution: New Drug—Limited
by Federal (or United States) law to investigational use”—
should be on the clinical site pharmacy label.
16
All labeling
must also comply with applicable local and state regulations.
In addition, the clinical research pharmacist should ensure
that any required medication counseling (e.g., for psychiat-
ric or teratogenic medications) is to be provided by the clini-
cal research pharmacist or an appropriate member of the
clinical site study team.
Considerations for Blinded Studies
When preparing dispensing guidelines for blinded studies
in which the clinical research pharmacy staff are unblinded,
the clinical research pharmacist should be cognizant that
there are clinical site personnel with direct patient care re-
sponsibilities who are blinded to the research participant’s
treatment assignment, and the dispensing guidelines should
be designed accordingly. Ideally, unblinded clinical site
personnel should not be involved in direct patient care for
study participants. Interactions between blinded and un-
blinded personnel should be minimized, and care should
be taken to avoid communication that could inadvertently
reveal a participant’s treatment assignment. Any pharmacy
staff member who is unblinded must use extreme care when
communicating with blinded staff regarding any patient care

Research–Guidelines 649
information in order to avoid revealing the participant’s
blinded treatment assignment.
Active and placebo investigational drug products must
be identical in appearance, labeling, preparation time, expi-
ration date and time, and supplies used. Considerations when
preparing blinded investigational drug products include
•
Ensuring that the preparation times for the active drug
and the placebo dose are the same.
•
Providing the identical information on the active drug
and placebo labels, including the expiration date and
time; if the active drug and placebo products have dif-
ferent expiration dates and times, the shorter of the 2
times should be used.
•
Making the port on intravenous bags look the same
(i.e., if a commercial bag of diluent is used as the
placebo, insert a needle into the port using aseptic
technique to give the appearance that a drug has been
added; if additive port covers are used, ensure match-
ing covers).
•
Using the same type of needle and tubing for adminis-
tration of the active drug and the placebo dose.
An unblinding process must be established that allows the
blinded treatment assignment to be determined. Examples of
events that require unblinding include a research participant
experiencing a severe adverse reaction and an unauthorized
person ingesting or being administered the investigational
drug product. An unblinding process that allows for immedi-
ate determination of the participant’s treatment assignment
or access to the sponsor’s medical monitor should be devel-
oped. In order to allow continuous access to the unblinding
process in the event of an emergent need to determine a par-
ticipant’s treatment assignment, a clinical site-specific plan
should be developed that is not dependent solely on the PI.
When labeling a blinded study medication, the clinical
research pharmacy must label it such that the blinding is pro-
tected and that clinical site study staff are not able to deter-
mine the actual contents of the container. The product should
be labeled as “drug name or placebo.” The protocol acronym
should not be used solely in place of the product name; if a
research participant presents at an emergency department,
the treating physician should be able to identify the potential
contents of the container so that treatment is not delayed.
Barcoding of Investigational
Drug Products
Improved patient safety with barcoding has been well docu-
mented.
17
Although barcoding is desirable, the lack of a
standard system limits its ability to be implemented with
investigational drug products. Development of a standard-
ized barcode lexicon that will facilitate the implementation
of a global barcoding system is necessary. ASHP has urged
the Food and Drug Administration (FDA) and other regula-
tory agencies, standard-setting bodies, contracting entities,
health systems, and pharmaceutical manufacturers to de-
velop and implement a universal symbology (e.g., barcodes,
radio frequency identifiers) that are readily deciphered by
commonly used scanning equipment to code for the National
Drug Code, lot number, and expiration date on all unit dose,
unit-of-use, and injectable drug packaging.
18
Investigational Drug Product
Accountability and Documentation
Detailed records, required to be kept by the sponsor, must
identify the investigator to whom the investigational drug
product is shipped as well as the date, quantity, and batch or
code mark of such shipment.
19
The clinical site is required to
maintain records detailing the participant to whom the inves-
tigational drug product was dispensed, the date, the quantity,
and the batch or code mark dispensed. Incomplete or inac-
curate drug accountability is a deficiency frequently cited in
Form FDA 483 Inspectional Observations notices.
20
U.S. law
does not require the use of an expiration date, use-by date,
or retest date on product labels. In many cases, the date may
be found on the packing slip; however, the clinical research
pharmacy may reach out to the sponsor to request documen-
tation of the retest or expiration date.
Routine inventory counts (e.g., monthly) should be
performed for each investigational drug product in order to
ensure that the physical quantity on hand corresponds to the
quantities recorded on the drug accountability record form
(DARF) and to manage investigational drug product with
limited use dates. Any discrepancy should be reviewed and
resolved for each investigational drug product.
According to the CFR and GCP guidelines,
1,2
inves-
tigational drug product receipt, dispensing, participant re-
turns, and disposition are required transactions that must
be documented. Sponsors may provide investigational
drug product accountability records, or individual clinical
research pharmacies may have their own accountability re-
cords. Frequently, sponsor-provided DARFs do not contain
sections to document all of the required transactions. For
clinical research pharmacies with multiple ongoing clinical
studies, DARFs from different sponsors requiring inconsis-
tent information can cause confusion. Use of standardized
DARFs containing sections for the required transactions
would ensure consistency and compliance with applicable
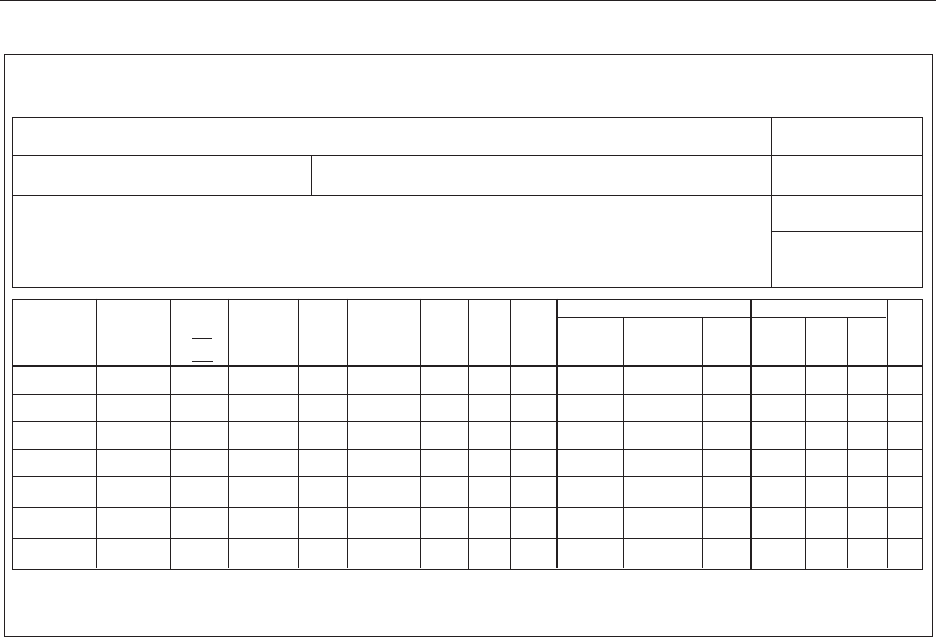
regulations. Figure 1 displays a template for a DARF, which
can be customized for different clinical protocols and sites
but maintains a standard appearance that will allow clini-
cal research pharmacy personnel to complete the forms cor-
rectly and provide sponsors with the required documenta-
tion. The form allows the site to document the receipt and
dispensing of individual bottles, vials, or kits, and the header
of the form can be customized for the specific needs of the
protocol. If a hard copy of the DARF is required by sponsors
and an electronic dispensing/accountability system is used,
it must have the capability of printing a hard copy of the
DARF that shows the required transactions. At a minimum,
the items on the form should be adhered to, ensuring site and
sponsor compliance with regulatory agencies. The clinical
research pharmacy DARF may be customized to include ad-
ditional information requested by sponsors, such as the time
of dose preparation or investigational drug product container
numbers.
When the investigational drug product is transferred
from the clinical research pharmacy to a satellite pharmacy
or dispensing location, a separate DARF must be main-
tained with the investigational drug product. The DARF in
the clinical research pharmacy should reflect the transfer of
investigational drug product to the satellite location, and the
satellite location’s DARF should reflect receipt of the inves-
tigational drug product from the clinical research pharmacy.
650 Research–Guidelines
The satellite location’s DARF should reflect all dispensing
activity at that location. Any remaining investigational drug
product must be returned to the clinical research pharmacy
and documented in both DARFs accordingly.
TheDARFisanocialrecord,and,assuch,anycor-
rections that are made must be performed by making a
line through the
original entry, accompanied by the re-
corder’s initials and date. The original entry may not be
obscured.
21
Investigational Drug Product Receipt
The clinical protocol or investigational drug product han-
dling manual will indicate how to obtain the investigational
drug product once a study site is approved to receive the
product from the sponsor. The study site may need to enroll
the first research participant before the product is shipped,
or a study site may be allowed to have the investigational
drug product onsite in anticipation of participant enrollment.
When the product is shipped, the sponsor must ensure it is
appropriately labeled so that it is easily identified as an in-
vestigational drug product for a specific clinical protocol.
The immediate packaging container for the investigational
drug product is required to bear the statement “Caution:
New Drug—Limited by Federal (or United States) law to
investigational use.”
16
In addition, it is strongly recommended that the spon-
sor’s investigational drug product label contain the follow-
ing information
22
:
•
Investigational drug product name
•
Investigational drug product name may be the
generic name, company molecule name, or com-
pound name but must match the name used in
the clinical protocol; the sole use of protocol ac-
ronyms should be avoided.
•
If the investigational drug product used in the
clinical research study is blinded and placebo con-
trolled, the label must indicate “[Investigational
Drug Product Name] or Placebo.”
•
Investigational drug product strength or concentration
unless this aspect of the trial is blinded
•
Investigational drug product quantity (e.g., number of
tablets, volume)
•
Investigational drug product lot number and/or con-
tainer or kit number
•
Lot number(s) should be referenced in the pack-
ing slip or receipt document, and terminology
should be consistent with the clinical research
study-related documentation (i.e., if the lot num-
ber is labeled as “Lot Number” on the package,
it should not be referred to as “Batch Number” in
supporting documentation).
•
Investigational drug products may also contain
a unique container or kit number that will be
used to identify the contents or to assign specific
container or kit to a participant; in that case, the
container or kit number may appear in place of
the lot number, but the container or kit number
must be cross-referenced with the packing slip.
•
Expiration or retest date (period of use) of the investi-
gational drug product
Figure 1. Template of a drug accountability record form. PI = principal investigator, CRA = clinical research associate.
(site name) Clinical Research Pharmacy
Drug Accountability Record Form (DARF)
Protocol Title: Internal Protocol #:
PI: Source: Lot:
Kit:
Exp/Retest:
Study Drug Name:
Synonym:
Str
ength:
Dosage Form:
Units Per Container:
Drug Location:
Drug Storage:
Room T
emp ___ Refrig # ___ Other ____
Receipt
Or
Dispense
Date
dd/mmm/yyyy
Rx# or
Pt Care Unit
Patient
Initials
FML
or
F - L
Patient Study
#
Dose/
Frequency
Quantity
Received or
Dispensed
Balance Staff
Init
CRA
Verify
Init
Patient Returns
# Returned Date Returned Staff
Init
Disposition
Return to
Sponsor
or Destroy
Date Staff
Init
CRA
Verify
Init
Comments: ___________________________________________________________________________ Page ____ of ____

Research–Guidelines 651
•
While it is preferred to be on the investigational
drug product label, this information may be pro-
vided on the packing slip or via a memo included
with the shipment to the clinical study site.
•
Sponsor or manufacturer name and address
•
Clinical research protocol number
•
When a sponsor is working on clinical studies
using the same investigational drug product in
parallel protocols for different indications, it
may be difficult for a clinical site to determine
which protocol the product belongs to unless this
information is provided on the container.
•
The sponsor may provide pooled supplies of in-
vestigational drug products to be used for mul-
tiple clinical research studies at the same clini-
cal study site. The investigational drug product
may be labeled with multiple clinical research
protocol numbers, or the clinical research phar-
macy may be required to assign each individual
unit to a specific clinical research study at the
time of dispensing. The assignment process must
be clearly explained in the investigational drug
product handling manual or the clinical protocol
and be communicated clearly to the clinical re-
search pharmacies.
•
Oral medication intended to be dispensed to a partici-
pant for self-administration at home must comply with
the Federal Poison Prevention Packaging Act
23
and be
packaged in a child-resistant container. If the disease
indicated in the clinical protocol is not conducive to
this type of packaging, a statement must be included in
the consent form alerting the potential participant that
there is a risk to children in the home due to the lack of
child-resistant packaging.
The investigational drug product shipment must be ac-
companied by a packing slip. Clinical research pharmacy
staff are responsible for verifying the contents against the
packing slip and assessing the condition of the package.
Proof of receipt should be provided to the sponsor or sup-
plier; this should be clearly outlined in the protocol or
investigational drug product handling manual or on the
packing slip. Any discrepancies (e.g., broken vials, miss-
ing product, temperature excursions) must be reported
immediately, and the resolution of discrepancies must be
appropriately documented. Sponsors may request that the
product be quarantined in the interim; quarantined product
must be maintained under the specified storage conditions.
Sponsors should be prompt in responding to discrepancies
or temperature excursions. The packing slip, shipment tem-
perature records, and any other documents received with
the investigational product shipment shall be stored in
the pharmacy study-specific file. All investigational drug
products received must be documented on a DARF.
Investigational Drug Product Dose
Preparation and Dispensing
When preparing and dispensing an investigational drug
product, the clinical research pharmacy should utilize site-
developed, study-specific dispensing guidelines to ensure
the investigational drug product handling requirements in
the protocol are met. The clinical research pharmacy may
store partial or empty vials of nonhazardous investigational
drug product in a limited-access, secure area until returned
or destroyed per the sponsor’s direction. Sponsors should be
diligent in the reconciliation and disposition of the used ma-
terials. Used and partial vials of hazardous investigational
drug product should be destroyed according to the institu-
tional policies and procedures immediately after dose prepa-
ration.
Remote Site or Clinic Dispensing
Remote site or clinic dispensing within a health system may
be considered when clinical research pharmacy involvement
may present a hardship for research participants or adversely
affect the conduct of the clinical research study, especially
in situations where timely access to the investigational drug
product may be a challenge.
The clinical research pharmacy should review studies
that may require remote site or clinic dispensing before IRB
review to assist in determining how the investigational drug
product should be managed at the remote site or clinic and
in determining the best dispensing option. In situations in
which pharmacist dispensing is not practical, physician dis-
pensing may be an option. State laws and regulations should
be reviewed to determine whether physician dispensing is
permitted. If physician dispensing is allowed, all applicable
state and federal laws and regulations with respect to han-
dling investigational drug products (e.g., secure storage con-
ditions, labeling requirements for outpatient use) as well as
those regulating accountability and dispensing must be fol-
lowed. The clinical research pharmacy should provide guid-
ance on the applicable state laws and regulations that apply.
In such cases, the PI must maintain appropriate study-
specific accountability records. These records should docu-
ment the investigational drug product received and dis-
pensed to participants enrolled in the clinical research study.
Investigational drug product returned from participants and
final disposition of investigational drug product must also
be recorded.
The clinical research pharmacy should also perform
periodic audits and inspections of storage facilities and drug
accountability procedures that are managed by the PI.
24
Investigational Drug Product Returned
from Participants
Per GCP, the PI is responsible for ensuring and assessing
clinical research protocol adherence.
2
This may include
receiving, counting, and documenting investigational drug
product returned from participants in the case report form or
the electronic medication administration record, if required,
in the protocol. This function may be delegated by the PI
to the clinical research pharmacy; in such cases, the unused
supply and empty investigational drug product containers
should be returned to the clinical research pharmacy for
documentation of the unused investigational drug product
returns from participants and its final disposition. The clini-
cal research pharmacy should record on the DARF the date
and the quantity of investigational drug product returned, as
directed in the investigational drug product handling man-
ual. On occasion, the date of the investigational drug product

652 Research–Guidelines
count may differ from the actual date of participant return
because the clinical research pharmacy may not be able to
process the return immediately. Returned investigational
drug product should be deemed not usable and should be
stored in an area separate from the investigational drug prod-
uct that is available for dispensing to research participants.
Used or partially used nonhazardous investigational
drug product returns should be stored by the clinical re-
search pharmacy in a limited-access, secure area until di-
rected by the sponsor to return or destroy the product. The
clinical research pharmacy should determine storage space
availability and notify the sponsors in advance of any space
limitations. The sponsor should visit the clinical research
pharmacy at appropriate intervals based on study activity to
return or destroy used investigational drug product. If such
visits do not occur, the clinical research pharmacy should
contact the sponsor to obtain permission to destroy or return
the product.
Hazardous investigational drug product returns and
returns of investigational drug product supplied by the
National Cancer Institute should not be stored onsite. These
products should be destroyed under the appropriate institu-
tional hazardous drug disposal procedures immediately af-
ter counting and documentation on the DARF; exceptions
should require approval by the clinical research pharmacist
and the PI and should be stated in the dispensing guidelines.
Each site should refer to its specific institutional policies re-
garding the destruction of hazardous drug products.
Investigational Drug Product
Final Disposition
For investigational drug product returned to the sponsor or
destroyed onsite, the return or destruction must be docu-
mented on the DARF by the clinical research pharmacy.
If required by the sponsor, a separate return or destruction
form may be used to document return or destruction of the
investigational drug product. If the return of unused inves-
tigational drug product to the sponsor is required per the
clinical research protocol, instructions on the return proce-
dure should be provided to the clinical research pharmacy
staff by the sponsor. If destruction of investigational drug
product by the clinical research pharmacy is approved by
the sponsor, the product should be destroyed per institu-
tional policy. If the sponsor requests return of a hazardous
investigational drug product, the shipper must comply with
U.S. Department of Transportation regulations for ship-
ping hazardous material.
25
Investigational drug products
that are controlled substances should be returned to the
sponsor for final disposition or destroyed per institutional
policy; returns of such drug products must adhere to Drug
Enforcement Administration regulations for shipping con-
trolled substances.
26
Clinical Research Pharmacy Study File
The clinical research pharmacy should create a study-spe-
cific pharmacy file using selected essential documents that
are deemed necessary by the clinical research pharmacist
for the routine management of investigational drug prod-
ucts. Original documents (e.g., DARFs, investigational drug
product physician orders, packing slips) that are part of the
clinical research study-specific pharmacy file are consid-
ered source documentation. These essential clinical research
pharmacy documents will be retained throughout the study
and must be readily available for inspection by the spon-
sor (or designee), PI (or designee), and regulatory agencies
(e.g., FDA). Copies of temperature monitoring logs and pol-
icies do not need to be filed in individual clinical research-
specific files but should be available on request.
Documents in the pharmacy file may be stored in
hard copy or electronic format and should include sponsor-
provided and site-prepared documents. Sponsor-provided
documents should include the following:
•
IRB-approved clinical research protocol and associ-
ated amendments
•
Investigator’s brochure and associated amendments
•
Investigational drug product handling manual (version
controlled) and associated amendments
•
Investigational drug product ordering instructions and
forms
•
IRT (IVRS/IWRS) manuals (instructions and forms)
•
Investigational drug product shipment documentation
(e.g., packing slips, other drug receipt documents)
•
Supporting investigational drug product information
(e.g., package insert, safety data sheet, certificate of
analysis, as applicable)
•
Expiration and retest correspondence (period of use)
•
Temperature excursion forms (if required)
•
Correspondence (e.g., communication sheet, letters, e-
mails)
•
Investigational drug product disposition information
and forms
•
Miscellaneous documents (e.g., worksheets, sponsor-
specific forms)
Site-prepared documents should include the following:
•
IRB approval letter
•
Access to IRB-approved consent (to establish docu-
mentation of research participant-specific medication
training disclosure)
•
Pharmacy-prepared dispensing guidelines
•
Pharmacy fee sheet with billing information
•
Pharmacy computer system entry information
•
Investigational drug product physician order or pre-
scription template(s)
•
List of authorized prescribers for the clinical trial
•
Research participant list and treatment assignment (if
applicable)
•
Clinical research pharmacy and pharmacy satellite
DARFs (if applicable)
•
Monitoring visit and audit forms
•
Notes to file
If an investigational drug product is not dispensed from the
clinical research pharmacy, a satellite-specific file should
be created that includes a research participant list, dispens-
ing guidelines, DARFs, and other study-specific documents
(e.g., investigational drug product dose calculation work-
sheets); the file should be kept in the satellite pharmacy
where the investigational drug product is being dispensed.

Research–Guidelines 653
Monitoring Visits or Audits of the Clinical
Research Pharmacy
It is the responsibility of the sponsor to monitor the prog-
ress of the clinical research study to ensure data integrity
and participant safety.
1
If the clinical research study is an
investigator-initiated protocol (sponsor–investigator), it is
the responsibility of the local PI to meet all of the require-
ments of the sponsor, including monitoring and safety-
reporting regulations.
The sponsor may delegate the monitoring and auditing
responsibilities to a contract research organization. Clinical
research associates who are employees of the sponsor or
their agents from a contract research organization will be
sent to monitor the clinical site. Sponsor audits at clinical
sites may be conducted by representatives of the sponsor or
independent auditors as well as inspectors from regulatory
authorities. Communication regarding clinical site monitor-
ing expectations will occur during the site initiation visit.
The clinical research associate (monitor) will visit
the clinical study site to review the records and verify the
accuracy of the documentation with the source documents
onsite. Source documents include any original documents,
data, and records, including (but not limited to) patient
medical rec ords, visit notes, protocol data collection forms,
laboratory notes, participant drug administration diaries,
and the clinical research pharmacy DARFs and associated
records. The monitor must have access to the source docu-
ments; in some cases, this may require access to electronic
medical records. Access to these systems should be pro-
vided in accordance with institutional policies.
The monitor should communicate with the clinical
study site to determine a mutually agreeable time to meet
with study staff and reserve a space to work. The clinical
site should accommodate these visits whenever possible.
The clinical study site should have a dedicated space in the
same area as the protocol records where the monitor can
work. The PI must be available for questions and discussion
at some time during the visit. Some research protocols will
allow remote monitoring activities in lieu of a visit to the
site, and monitors may send queries to the clinical site for
clarification. Depending on the time required by the clinical
research pharmacy staff to respond to information requests,
an additional charge may be imposed to accommodate these
remote visits.
The monitor will need to visit the clinical research
pharmacy to review pharmacy documentation. If the clini-
cal research protocol is a blinded study and the clinical re-
search pharmacist is unblinded, there should be a separate
unblinded monitor specifically for clinical research phar-
macy review. The unblinded contact(s) should be clearly
stated in the pharmacy records. All others should be con-
sidered blinded and communications handled accordingly.
An appointment should be made for each clinical research
pharmacy visit to allow the staff to adequately prepare.
27
The clinical research pharmacy staff should meet with the
monitor and provide access to pharmacy source documents
for review. The monitor should confirm drug account-
ability and storage conditions and review a participant’s
returned investigational drug product. The monitor may
return investigational drug product to the sponsor or au-
thorize its destruction. All personal health information of
the participants should be removed from investigational
product labels and forms before being returned to the
sponsor or should be destroyed.
Monitoring Visit Logs
The clinical research pharmacy staff should docu-
ment monitoring visits and the reason for the meeting.
Information documenting the identity of the monitor,
company affiliation, study reviewed, and the duration of
the visit to the clinical research pharmacy is useful to the
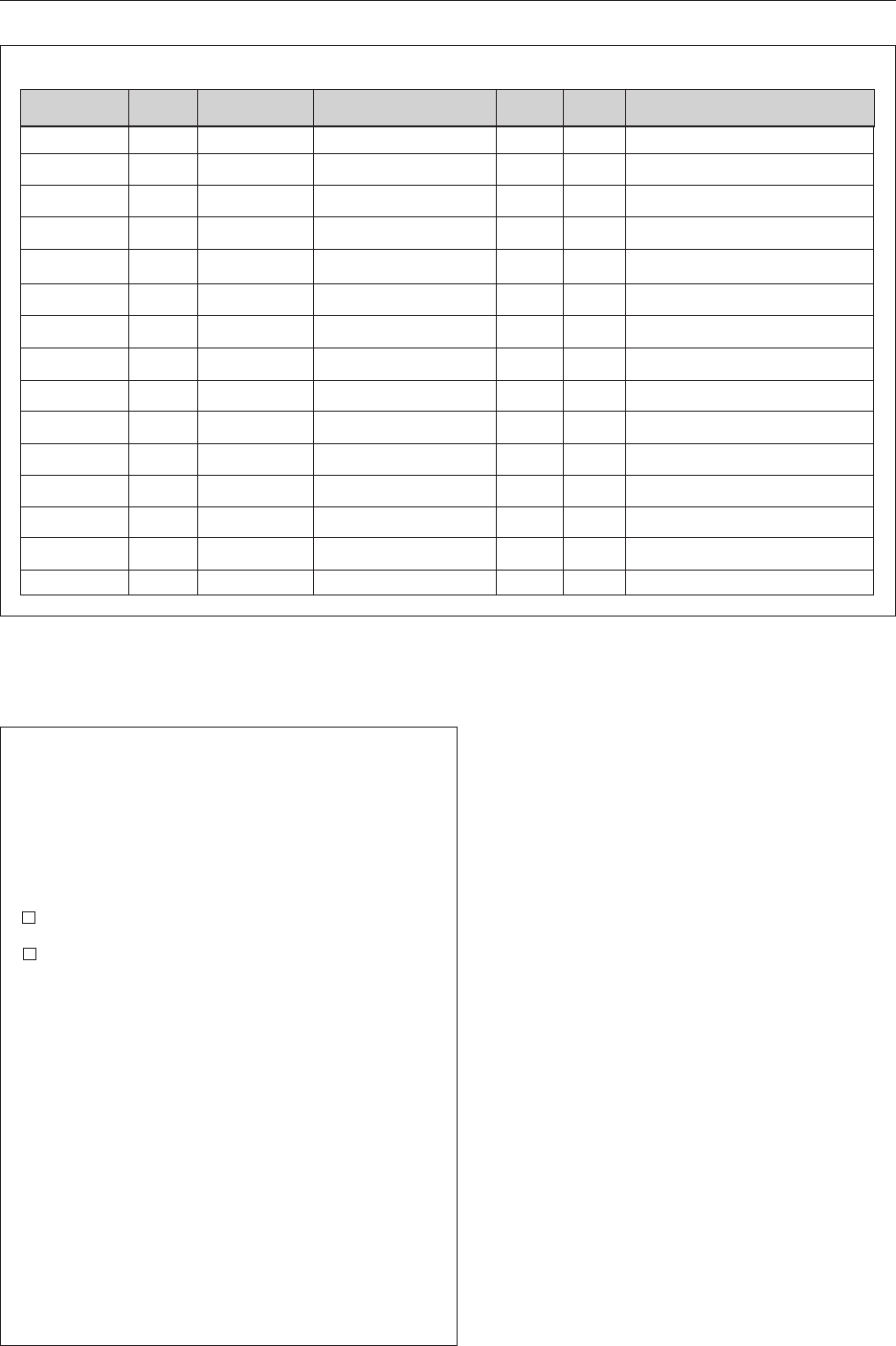
sponsor and the pharmacy department. Figure 2 provides
an IDS monitor/visitor log template to capture such infor-
mation.
At the end of a monitoring visit, the clinical research
pharmacy staff or the sponsor should document any out-
standing requests or issues that need to be completed before
the next visit. A template of a study monitor exit summary
report that can be used to capture issues for follow-up ap-
pears in Figure 3. Both the clinical research pharmacist and
the monitor should sign the form to document their under-
standing of requests.
Study Close and Archiving of the Clinical
Research Pharmacy Study Files
A site is required to retain all rec ords for at least 2 years after
the marketing application is approved for the indication be-
ing investigated. If the drug is not approved or if no applica-
tion is filed, then records must be retained for 2 years after
the investigation is discontinued.
26
Many institutions have
retention policies that exceed these requirements. Trials tak-
ing place in other countries may also be subject to longer
retention periods. Sponsors should indicate when this is the
case and follow retention requirements where applicable. It
is the responsibility of the sponsor to notify the clinical site
and the clinical research pharmacy of these requirements.
Each site must be familiar with state, local, and institutional
policies and regulations regarding record retention. The clin-
ical research pharmacy should provide written notice to the
sponsor of impending document destruction.
Upon termination of a study, the clinical research phar-
macy should work with the PI to determine who will main-
tain the following pharmacy-related documents:
•
Drug accountability records and documentation of in-
vestigational drug product destruction
•
Investigational drug product shipment documen-
tation
•
Proof of receipt and/or packing list
•
Certificates of analysis
•
Safety data sheets
•
Participant enrollment log
•
Participant identification code list
•
Participant-specific preparation records (batch
control records for compounded items) and
worksheets
•
Certain pharmacy documents, such as
filled prescriptions or medication orders,
must remain with the clinical research
pharmacy in accordance with state board
of pharmacy rules and regulations.
•
IRT-related documents
654 Research–Guidelines
Figure 3. Study monitor exit summary report template. CRP = clinical
research pharmacy.
Clinical Research Pharmacy
Study Monitor Exit Summary Report
Internal Protocol#__________
No Clinical Research Pharmacy action required
Follow-up Clinical Research Pharmacy action required
Study Monitor/Date
Contact Information:
CRP Staff/Date
(R.Ph. required for follow-up)
Figure 2. Template of an investigatioinal drug service monitor/visitor log.
Clinical Research Pharmacy Monitor/Visitor Log Month ____________ Page ____________
DATE Internal
Protocol #
DRUG/STUDY NAME TIME IN TIME OUT REASON FOR VISIT
•
Instructions for handling investigational drug product
•
Site-specific dispensing guidelines
•
Signature sheet and pharmacy delegation log
•
Sponsor investigational drug product and pharmacy-
related correspondence
Nonessential pharmacy documents (e.g., protocols,
amendments, investigator’s brochure, IRB correspondence)
shall be stored by the PI in the investigator study file. The
clinical research pharmacy does not need to maintain copies
of these documents.
The clinical research pharmacy files for terminated
studies that are not returned to the PI should be maintained
onsite, as space allows, and then moved to offsite, long-term
storage as needed and if available. The clinical research
pharmacy should maintain an onsite record of location for
retrieval from long-term storage. Records must be readily
retrievable in the event of an audit.
Clinical Research Pharmacists
as IRB Members
IRB members must disclose any actual and perceived con-
flicts of interest throughout their membership term and must
file a financial disclosure with the IRB office. This informa-
tion should be considered during assignment of IRB submis-
sions for review to avoid conflicts of interest.
An IRB member may be involved in the conduct of a
research study that solely involves the provision of a service
to a study (e.g., a pharmacist from the clinical research phar-
macy who prepares and dispenses study medications, a radi-

Research–Guidelines 655
ologist who performs diagnostic imaging that is part of the
research). In these cases, the IRB should not consider this a
conflict of interest with regard to reviewing an IRB submis-
sion, provided the member’s role in the study is limited to
the provision of a service to the PI and he or she is not oth-
erwise engaged in the study. Under these circumstances, the
clinical research pharmacist serving on an IRB should not
be listed on the Statement of Investigator (Form FDA 1572).
Summary
Clinical study sites that handle a significant volume of clini-
cal studies should establish a formal IDS pharmacy utiliz-
ing these best practices as a foundation. Standardizing the
processes for managing investigational drug products used
by clinical research pharmacies will improve patient safety
and protect study data integrity while maintaining regulatory
compliance. Standardization will also provide guidance for
clinical study sites that are new to handling investigational
drug products. Adopting these best practices will enable
clinical research pharmacies to enhance their collaboration
with sponsors and the biopharmaceutical industry by align-
ing processes and systems to improve clinical study execu-
tion. Ultimately, these best practices will support the devel-
opment of new and innovative medications for the patients
who need them most.
References
1. Investigational New Drug Application, 21 CFR 312.
www.ecfr.gov/cgi-bin/text-idx?SID=0261b7eecf94ba
de1f78efd3b22ee7f6&mc=true&node=pt21.5.312&rg
n=div5 (accessed 2017 Jun 19).
2. International Conference on Harmonisation
of Technical Requirements for Registration of
Pharmaceuticals for Human Use. ICH harmonized tri-
partite guideline: guideline for good clinical practice.
J Postgrad Med. 2001; 47:45–50.
3. Current Good Manufacturing Practice for Finished
Pharmaceuticals (21 CFR 211). www.ecfr.gov/cgi-bin/
text-idx?SID=0261b7eecf94bade1f78efd3b22ee7f6&
mc=true&node=pt21.4.211&rgn=div5 (accessed 2017
Jun 19).
4. Drug Amendments of 1962. Pub. L. 87-781, Oct. 10,
1962, 76 Stat. 780 (21 U.S.C. 301). http://uscode.
house.gov/table3/87_781.htm (accessed 2017 Jun 19).
5. DiMasi JA, Grabowski HG, Hansen RW. Cost of
developing a new drug (November 2014). http://csdd.
tufts.edu/files/uploads/Tufts_CSDD_briefing_on_
RD_cost_study_-_Nov_18,_2014..pdf (accessed 2017
Jun 19).
6. Rough SS, McDaniel M, Rinehart JR. Effective use of
workload and productivity monitoring tools in health-
system pharmacy, part 1. Am J Health-Syst Pharm.
2010; 67:300–11.
7. Rough SS, McDaniel M, Rinehart JR. Effective use of
workload and productivity monitoring tools in health-
system pharmacy, part 2. Am J Health-Syst Pharm.
2010; 67:380–8.
8. Health Insurance Portability and Accountability Act
of 1996 (Pub. L. 104-191). http://uscode.house.gov/
table3/104_191.htm (accessed 2017 Jun 19).
9. Protection of Human Research Subjects (45 CFR 46). www.
hhs.gov/ohrp/regulations-and-policy/regulations/45-
cfr-46/index.html (accessed 2017 Jun 19).
10. National Commission for the Protection of Human
Subjects of Biomedical and Behavioral Research.
Ethical principles and guidelines for the protection of
human subjects of research (April 18, 1979). www.
hhs.gov/ohrp/regulations-and-policy/belmont-report/
index.html (accessed 2017 Jun 19).
11. General responsibilities of sponsors (21 CFR 312.50).
www.ecfr.gov/cgi-bin/text-idx?SID=10f9559cb05469
f95eb97b446651b615&mc=true&node=se21.5.312_1
50&rgn=div8 (accessed 2017 Jun 20).
12. The United States pharmacopeia, 36th rev., and The
national formulary, 31st ed. Rockville, MD: United
States Pharmacopeial Convention; 2013:11.
13. National Institute of Standards and Technology. Good
measurement practice for assignment and adjustment of
calibration intervals for laboratory standards (January
2016). www.nist.gov/sites/default/files/documents/
pml/wmd/labmetrology/GMP_11_20160121.pdf
(accessed 2017 Jun 19).
14. Food and Drug Administration. Information sheet
guidance for sponsors, clinical investigators, and
IRBs: frequently asked questions—statement of in-
vestigator, Form FDA 1572 (May 2010). www.fda.
gov/downloads/RegulatoryInformation/Guidances/
UCM214282 (accessed 2017 Jun 19).
15. Siden R, Tamer HR, Skyles AJ et al. Pharmacist-
prepared dispensing guidelines for drugs used in clinical
research. Am J Health-Syst Pharm. 2012; 69:1021–6.
16. General responsibilities of investigators (21 CFR
312.60). www.ecfr.gov/cgi-bin/text-idx?SID=10f955
9cb05469f95eb97b446651b615&mc=true&node=se2
1.5.312_160&rgn=div8 (accessed 2017 Jun 19).
17. Poon EG, Keohane CA, Yoon CS, et al. Effect of bar-
code technology on the safety of medication adminis-
tration. N Engl J Med. 2010; 362:1698–707.
18. American Society of Health-System Pharmacists.
ASHP statement on barcode-enabled medication ad-
ministration technology. Am J Health-Syst Pharm.
2009; 66:588–90.
19. Recordkeeping and record retention (21 CFR 312.57).
www.ecfr.gov/cgi-bin/text-idx?SID=10f9559cb05469
f95eb97b446651b615&mc=true&node=se21.5.312_1
57&rgn=div8 (accessed 2017 Jun 19).
20. Office of Scientific Investigations. OSI metrics.
www.fda.gov/AboutFDA/CentersOffices/
OfficeofMedicalProductsandTobacco/CDER/
ucm256374.htm (accessed 2017 Jun 19).
21. National Cancer Institute. Pharmaceutical management
branch investigational drug accountability series. Drug
accountability record form (DARF) basics. http://
ctep.cancer.gov/branches/pmb/drug_training_videos/
DARF_Basics_Handout.pdf (accessed 2017 Jun 19).
22. Institute for Safe Medication Practices. Product-
related issues make error potential enormous with in-
vestigational drugs (November 1, 2007). www.ismp.
org/newsletters/acutecare/articles/20071101.asp (ac-
cessed 2017 Jun 19).
23. Poison Prevention Packaging Act of 1970 (15 U.S.C.
1471−17;Pub.L.91-601,84Stat.16).www.cpsc.gov/
s3fs-public/pppa.pdf (accessed 2017 Jun 19).

656 Research–Guidelines
24. Shehab N, Tamer HR. Investigational drugs not dis-
pensed by pharmacy: regulatory issues and the role
of the investigational drug service. Am J Health-Syst
Pharm. 2004; 61:1882–4.
25. Shippers—general requirements for shipments and
packagings (49 CFR 173). www.ecfr.gov/cgi-bin/text-
idx?SID=5b920076cbe059040feb2b85e9951775&m
c=true&tpl=/ecfrbrowse/Title49/49CIsubchapC.tpl
(accessed 2016 Jul 19).
26. Ultimate user delivery for the purpose of recall or
investigational use of drugs (21 CFR Part 1317.85).
www.ecfr.gov/cgi-bin/text-idx?SID=26cb45a08d1725
13244d104e129e0caf&mc=true&node=se21.9.1317_
185&rgn=div8 (accessed 2017 Jun 19).
27. Siden R, Tankanow RM, Tamar HR. Understanding
and preparing for clinical drug trial audits. Am J
Health-Syst Pharm. 2002; 59:2301–8.
These guidelines were developed through the ASHP Section
of Inpatient Practitioners and approved by the ASHP Board of
Directors on November 8, 2017.
Stephen C. Kay, B.S.Pharm., Clinical Research Pharmacy, Pfizer,
Andover, MA.
Darlette G. Luke, B.S.Pharm., Investigational Drug Service,
Fairview Health Services, University of Minnesota Medical Center,
Minneapolis, MN.
Helen R. Tamer, Pharm.D., Research Pharmacy, Michigan Medicine
(University of Michigan), Ann Arbor, MI.
ASHP gratefully acknowledges the following individuals for re-
viewing these guidelines (review does not imply endorsement):
Uzma Afzal, Pharm.D.; Beth McLendon Arvik, Pharm.D.; Molly
A. Camis, Pharm.D., BCPS; Scott Fields, Pharm.D.; Mark G.
Klang, Ph.D., M.S., BCNSP; Eimeira Padilla-Tolentino, Pharm.D.;
Michele Page, M.B.A.; John Petrich, M.S.; Marjorie Shaw Phillips,
M.S., FASHP; Kenneth Rockwell Jr., Pharm.D., M.S.; Erika L.
Thomas, M.B.A., B.S. Pharm.; and John Vetrano, Pharm.D.
The authors have declared no potential conflicts of interest.
Copyright © 2018, American Society of Health-System Pharmacists,
Inc. All rights reserved.
The bibliographic citation for this document is as follows: American
Society of Health-System Pharmacists. ASHP guidelines for the
management of investigational drug products. Am J Health-Syst
Pharm. 2018; 75:561–73.
